The ground cherry (Physalis pubescens), also known as golden strawberry, Chinese lantern, low ground cherry, hairy ground cherry, and husk tomato, is one of the most well-known species (El Sheikha, 2004; USDA Natural Resources Conservation Service, 2016). It is grown in tropical, subtropical, and temperate climates all over the world, including the United States, Mexico, Colombia, Egypt, Zimbabwe, Kenya, Madagascar, and South Africa, but it is especially popular in Northeast China for the reason of its delicious flavor, nutritional content, and prospective health welfares (Hao et al., 2017). Vitamin C, potassium, phosphorus, zinc, boron, polyphenols, and carotenoids are all found in abundance in P. pubescens. It also contains a good amount of important amino acids including valine, tryptophan and isoleucine, which is necessary for the health of human being (El Sheikha et al., 2010).
However, occurrence of bacterial leaf spot (BLS) disease on P. pubescens caused significant economic losses (Song et al., 2019). Xanthomonas euvesicatoria, X. vesicatoria, X. gardneri, and X. perforans are four the genospecies of Xanthomonas that have been associated to BLS disease (Jones et al., 2004; Kebede et al., 2014). It is a gram-negative rod-shaped bacterium using aerobic motility and a single polar flagellum. The host range of BLS included Solanum lycopersicum var. cerasiforme, Lycopersicon pimpinellifolium, S. lycopersicum, Capsicum annuum, C. anomalum, C. frutescens, C. baccatum, and C. chinensis (Baker et al., 2014; EFSA Panel on Plat Health, 2014).
Nevertheless, according to Song et al. (2019), P. pubescens was new host range in BLS caused by Xanthomonas euvesicatoria pv. euvesicatoria that affected the quality and quantity of the fruit. Furthermore, the leaf becomes circular water soaked lesions on the lower and upper epidermis in the early stages of infection, and the leaf spot gradually rises and then turns from white to dark brown until it reaches the necrotic stage with a chlorotic edges. Occasionally, leaf spots are covered with a black color and greasy appearance, and bacterial populations grow on nutrient agar resulting in yellow colonies, indicating the presence of Xanthomonas species (Song et al., 2019).
Comparative genomics is essential for obtaining information from biological sequences. Comparative genomics of pathogenic bacteria has helped to find out genetic determinants of pathogenicity and virulence, as well as provide insights into pathoadaptation evolution (Garita-Cambronero et al., 2018). Larrea-Sarmiento et al. (2018) performed 10 genomic comparative analysis of BLS pathogens (X. euvesicatoria, X. perforans, X. vesicatoria, and X. gardneri) with other BLS pathogens for genomic variation and target gene selection through average nucleotide identity (ANI) and BLAST comparison.
Gene selection is critical pneumonia for accurate evolution and identification of BLS pathogens. Accordingly, hrpB1 and hrpB2 colocalize in the periplasm and interact with hrcD, implying that they are part of the type 3-secretion systemŌĆÖs (T3SS) periplasmic substructure. X. euvesicatoria delivers effector proteins into host cells through the T3SS to stimulate disease and T3SS transforms an active catalytic serine/threonine protein kinase in plant cells, resulting in novel enzymatic activity. The T3SS, which releases effector proteins into host cells to weaken plant immunity and promote disease, is essential for Xanthomonas pathogenicity (Teper et al., 2016). The T3SS was necessary for reliable identification of the BLS pathogens (Larrea-Sarmiento et al., 2018). ATP-dependent DNA helicases RecQ is genome surveillance proteins that can be found throughout the animal kingdom. RecQ helicases are bacterial, fungal, animal, and plant genome surveillance proteins. RecQ family members are involved in gene targeting, DNA repair, and development (Mendonca et al., 1995). The identification of DNA repair genes in phytopathogens could aid in the understanding of the methods by which these organisms adapt to their surroundings, including plant infection (Martins-Pinheiro et al., 2004).
Further research into evaluating and detecting pathogens at the molecular level requires the selection of approaches that allow for easy and rapid detection. Since the 1990s, more laboratories have begun to use polymerase chain reaction (PCR) and its derivatives, such as digital PCR, multiplex PCR, nested-PCR, and quantitative PCR (qPCR), for pathogen recognition and identification of Xanthomonas species (Strayer et al., 2016; Yasuhara-Bell et al., 2017; Zhang and Tanner, 2017). Such approaches are typically highly specific, comparatively fast, and inexpensive, but they do have certain drawbacks, such as the need for specific equipment and skilled workers in most circumstances. New technologies based on isothermal amplification of DNA, such as, helicase-dependent amplification, recombinase polymerase amplification, and loop-mediated isothermal amplification (LAMP) have emerged in the first few years of this century, ability to overcome some of the disadvantages of PCR-based approaches (Larrea-Sarmiento et al., 2018; Zhong and Zhao, 2018).
In comparison to other molecular approaches, the LAMP method allows for the easy and rapid detection of pathogens in a short period. LAMP is a DNA polymerase with two interior primers, and two exterior primers, and the interior primer binds to its F2c or B2c primer on the objective DNA, while the exterior primer binds to its F3c or B3c primer, creating a single complementary sequence. At both ends of a DNA sequence, internal loop primers (LF and LB) have the ability to speed up and shorten the reaction time (Larrea-Sarmiento et al., 2018). Moreover, identification and pathogenicity characterization of Xanthomonas euvesicatoria pv. euvesicatoria reported on P. pubescens in China by concatenated phylogeny based on hrpB and four housekeeping genes (lepA-gyrB-gapA-gltA) (Song et al., 2019). Despite the occurrence of BLS outbreaks on P. pubescens in China, little study has been undertaken on the genetic characterization and distribution of the pathogenic Xanthomonas species on P. pubescens. The major goal of this study was to isolation and characterization of BLS strains isolated from P. pubescens in China and to develop genome wide comparison of 13 Xanthomonas spp. using NCBI whole genome sequence database to verify genomic diversification and showed site location of selected three genes (T3SS hrpB1 and hrpB2 genes and ATP-dependent DNA helicase recQ gene). Furthermore, to develop molecular detection methods from unique genomic sequences, identified by comparative genomics, which can distinguish different Xanthomonas pathotypes infecting P. pubescens. In addition, an evolutionary study was performed to ascertain the strainsŌĆÖ phylogenetic position within the Xanthomonas species.
Materials and Methods
Sample collection, isolation, and DNA extraction
Infected 52 specimens were taken from different areas of Heilongjiang Province, China, based on symptoms (circular water-soaked lesions, leaf spot with dark brown spot, and necrotic stage with chlorotic margin). The infected samples were cut into small pieces, dipped in 2% sodium hypochlorite, and then transferred to phosphate buffered saline for one day. They were then made into a suspension of 1 ├Ś 105 solution for the isolation of the bacterial strains cultured on nutrient agar in the Petri plate and incubated at 28┬░C for three days. A single colony was chosen and streaked numerous times on nutrient agar for pure culture. DNA was extracted from diseased and healthy plant material using the CTAB technique. This extracted DNA was kept at ŌłÆ80┬░C for subsequent analysis.
Whole genome sequence comparison, target selection, and gene location
Whole genome sequence data of 13 Xanthomonas strains were retrieved from NCBI GenBank database. The investigation of whole genome sequence comparison was carried out using binary methods: BLAST comparison and ANI. CGview Server assessed the blast comparison, while OrthoANI software (OAT) Lee et al. (2015) analyzed the ANI for more similarities and differences among the genomes. In whole genomes sequence analysis, X. euvesicatoria was compared with X. perforans, X. vesicatoria, X. gardneri, X. axonopodis pv. citrumelo, X. compestris pv. vesicatoria, X. arboricola pv. pruni, X. cucurbitae, and X. fragariae that casual agents of bacterial spot and other X. campestris pv. campestri, X. axonopodis pv. glycines, Xanthomonas translucens pv. translucens, and X. vasicola caused black rot, bacterial pustules and bacterial leaf streak diseases, respectively. Genomes of X. euvesicatoria (CP018467), X. axonopodis pv. citrumelo (CP002914), X. gardneri (CP018731), X. perforans (CP019725), X. campestris pv. vesicatoria (CP017190), and X. campestris pv. vesicatoria (AM039952) were aligned with progressive MAUVE for more compatibility among genomes and exact gene location of recQ gene in all genomes. CGview Server was also used to find the exclusive location of the ATP-dependent DNA helicase recQ gene in the X. euvesicatoria, which was used to construct specialized LAMP primers for the X. euvesicatoria.
Primer design, PCR protocol, and sequencing
Plant pathogen bacteria can be detected using PCR, which is a relatively easy and convenient approach. Three genes (hrpB1, hrpB2, and recQ) were chosen in this investigation for accurate identification of pathogenic strains X. euvesicatoria through PCR analysis. hrpB1 and hrpB2 primers were designed through Primer3 software, whereas recQ primer was designed by Primer Explorer V5 and additionally, F3 as a forward and B3 as a reverse primer used for PCR analysis (Table 1). Each PCR tube had a total of 25 ╬╝l of reaction, which contains 2 ╬╝l of template DNA and 0.4 ╬╝M, 0.4 ╬╝M, and 12.5 ╬╝l of forward primer, reverse primer, and 2├Ś Taq Master Mix, respectively (CoWin Biosciences, Beijing, China) and remaining volume cover with dd water. The cycling conditions were 3 min at 94┬░C, 35 cycles of denaturation at 94┬░C for 30 s, and annealing at 54┬░C for 30 s for both genes hrpB1 and hrpB2 whereas for recQ was at 53┬░C for 30 s, elongation at 72┬░C for 30 s, and a final elongation at 72┬░C for 10 min. PCR products were run on 1% agarose gel electrophoresis with 200 mA for 25 min. and then, PCR products sent to Sangon Biotech (Shanghai) Co., Ltd. (Shanghai, China) for sequencing.
Primer design for LAMP
To create specific LAMP primers for X. euvesicatoria, the ATP-dependent DNA helicase recQ gene was chosen. The specificity of this gene is important for genomic stability. Primer Explorer V5 software (https://primerexplorer.jp/e/) was used to construct sense and anti-sense primers of inner (FIP and BIP), outer (F3 and B3), and internal loop primers (LF and LB) (Table 1). Each primer was verified in silico using the BLASTn tool in the NCBI nucleotide database. The designed primers covered 100% of the query and were 100% identical to X. euvesicatoria.
Optimization of LAMP reaction
The evaluation of LAMP chemicals reaction was performed by optimization of reaction with different concentrations such as DNA template concentrations (10ŌłÆ1, 10ŌłÆ2, 10ŌłÆ3, 10ŌłÆ4, and 10ŌłÆ5), primers (1:2, 1:4, 1:8, and 1:12), MgSO4 (0, 4, 5, 6, 7, and 8 mM), time (15, 30, 45, and 60 min), temperature (57┬░C, 59┬░C, 61┬░C, 63┬░C, 65┬░C, 67┬░C, and 69┬░C), Bst (0.5, 1, 1.5, and 2), betaine (0.4, 0.6, 0.8, 1, 1.2, and 1.4 M), and hydroxynaphthol blue (HNB) for visual color change.
The final reaction mixture of 25 ╬╝l contains 2 ╬╝l of template DNA, primer (F3 and B3 as outer primer, FIP and BIP as inner primers), MgSO4, 1.4 mM of dNTPs, 1├Ś isothermal amplification buffer (containing 20 mM Tris-HCl, 10 mM (NH4)2SO4, 50 mM KCl, 2 mM MgSO4, and 1% Tween 20 with pH 8.8), 1 U of Bst 2.0 WarmStart DNA polymerase, betaine, and sterile double-distilled water. The experiment was carried out in a thermal water bath for the time between 15 to 60 min at a temperature between 57┬░C to 69┬░C. To confirm amplification, the amplified LAMP products 4 ╬╝l were examined on a 2% stained agarose gel with ethidium bromide.
Real-time PCR assay
The SYBR Green real-time PCR assay was used to test the infected samples of P. pubescens using target RecQ gene. iTaq Universal SYBR Green Supermix kit (Bio-Rad, Hercules, CA, USA) used for real-time PCR that contains dNTPs, iTaq DNA Polymerase, MgCl2, SYBR Green I, enhancers, stabilizers, and a blend of passive reference dyes. Each PCR reaction tube contained 10 ╬╝l of iTaq Universal SYBR Green Supermix kit and 0.4 ╬╝l of each 10 ╬╝mol/l primer, 1 ╬╝l template, and 8.6 ╬╝l ddH2O. Real-time PCR was performed with the following program: 2 min at 95┬░C, 30 cycles of denaturation at 95┬░C for 15 s, annealing at 54┬░C for 15 s, elongation at 72┬░C for 15 s, and a final elongation at 72┬░C for 2 min. DNA templates were replaced by double-distilled water as a negative control.
Phylogenetic tree analysis and model design
Phylogenetic trees were constructed using three unique target genes hrpB1, hrpB2, and recQ. PCR products were purified and sequenced by Sangon Biotech (Shanghai) Co., Ltd. All other sequenced data were retrieved from NCBI GenBank database and a basic alignment search technique (BLASTn) was used to match the unidentified sequences to sequences in public databases. All sequenced data were aligned through ClustalW integrated into MEGA 11 software. Evolutionary tree analysis was performed through maximum likelihood (ML) methods. In the evolutionary tree-building process, model selection is a critical phase. To estimate phylogeny, we employed standard DNA sequence evolution models. MAGA 11 was used to build 24 nucleotide substitution fit models.
Pathogenicity and specificity test of primers for recQ, hrpB1, and hrpB2 genes
On five cultivars of P. pubescens, bacterial isolates were tested for pathogenicity, and inoculation was done on seedlings that were five to six weeks old. The reference strains (Xeu1, Xeu2, and Xeu3) were cultured on nutritional agar (NA) medium in a Petri dish for three days at 28┬░C. Bacterial suspensions having 1 ├Ś 105 cfu/ml were prepared through 10-fold dilutions into nutrient broth medium, and spectrophotometrically adjusted suspensions 0.03 at 600 nm OD for inoculation.
Through needleless syringes, suspensions were injected into the anterior portion of the mesophyll of immature leaves and for control treatment, healthy leaf was used. Plants were placed into growth chamber with a photoperiod of 12 h and 28 ┬▒ 1┬░C and leaves were observed on daily bases. Re-isolation of infected leaves and growth on NA described above and in addition, extraction of DNA of infected leaves and non-template control (NTC) through CTAB method and performed PCR using primers of hrpB1, hrpB2, and recQ genes.
Furthermore, for the primer specification of hrpB1, hrpB2, and recQ, 52 strains were used including three reference strains (Xeu1, Xeu2, and Xeu3), two strains of Pseudomonas syringae pv. syringae (Pss1 and Pss2) and one strain of X. oryzae pv. oryzae (YH). These strains were collected from Plant Pathology Lab of Northeast Agricultural University and DNA was extracted through CTAB method. All these strains were observed through conventional PCR approach and after that run on 1% agarose electrophoresis gel with 200 mA for 25 min. Three strains, Xeu1, Xeu2, and Xeu3, were obtained from Song et al. (2019) for validation of X. euvesicatoria with recQ, hrpB1, and hrpB2 genes and these strains were applied to P. pubescens seedlings using the inoculation procedures described above.
The Canopeo app (https://canopeoapp.com) was used to calculate the leaf damage area by percentage at 3, 7, 14, and 21 days after inoculation to determine disease severity. Canopeo is a system-consuming RGB (red, green, and blue) programmer. The excess ratios of the B/G, R/G and green indexes were employed to construct pixel analysis. The results are represented in binary pictures, with white pixels indicating that the selection criteria were met (green canopy), and black pixels indicating that the selection criteria were not met (not green canopy) or that damage occurred (Patrignani and Ochsner, 2015). The percentage of leaves that have green color was used to determine severity; a score of 100% was connected with non-infected or healthy leaves, Xanthomonas infected leaves were associated with decreased percentages.
Results
Whole genome sequence comparison and selection of target gene
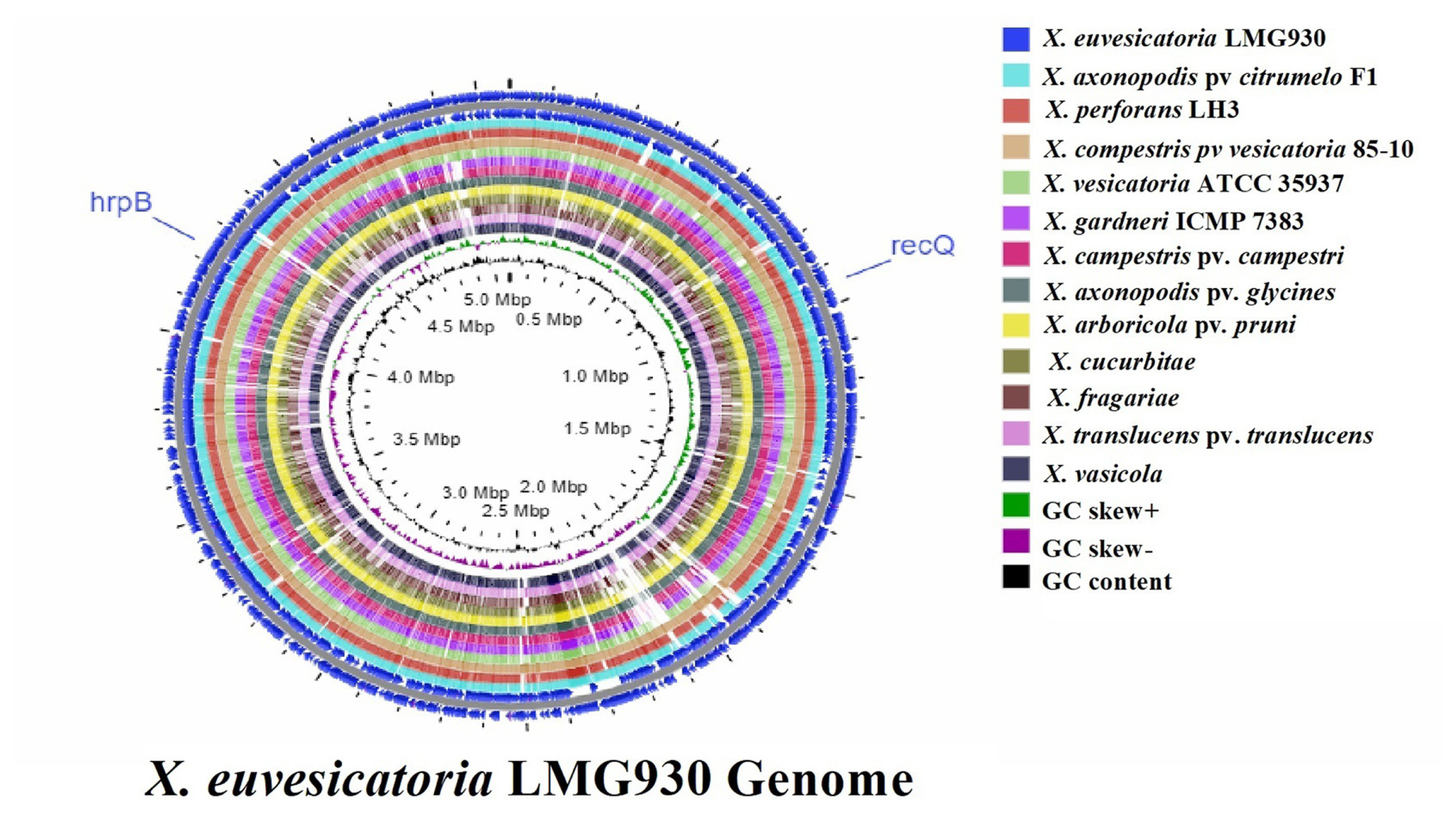
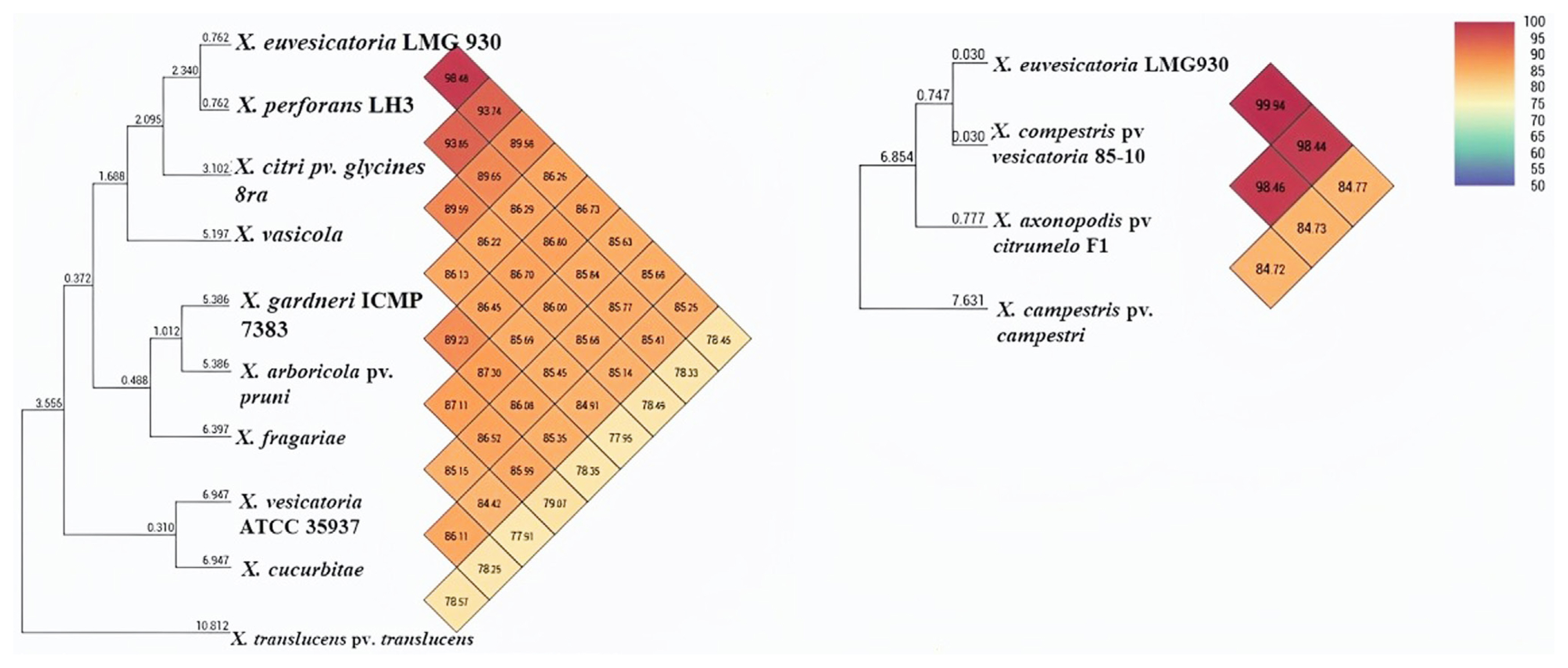
For more similarities or differences throughout 13 genomes, CGview Server was used to do whole genome sequence comparison analysis. The sequence data for whole genomes comparison was obtained from the NCBI GenBank genome. BLAST comparison and ANI are two extensively used methods for genomic comparison. Genome of X. euvesicatoria compared with other 12 genomes with the location of specific target gene recQ ATP-dependent DNA helicase and hrpB (hrpB1 and hrpB2) (Fig. 1). When compared to other genomes, the sequence of the reference genome X. euvesicatoria was closely related to the sequences of X. campestris pv. vesicatoria 85-10, X. axonopodis pv. citrumelo F1, and X. perforans LH3.
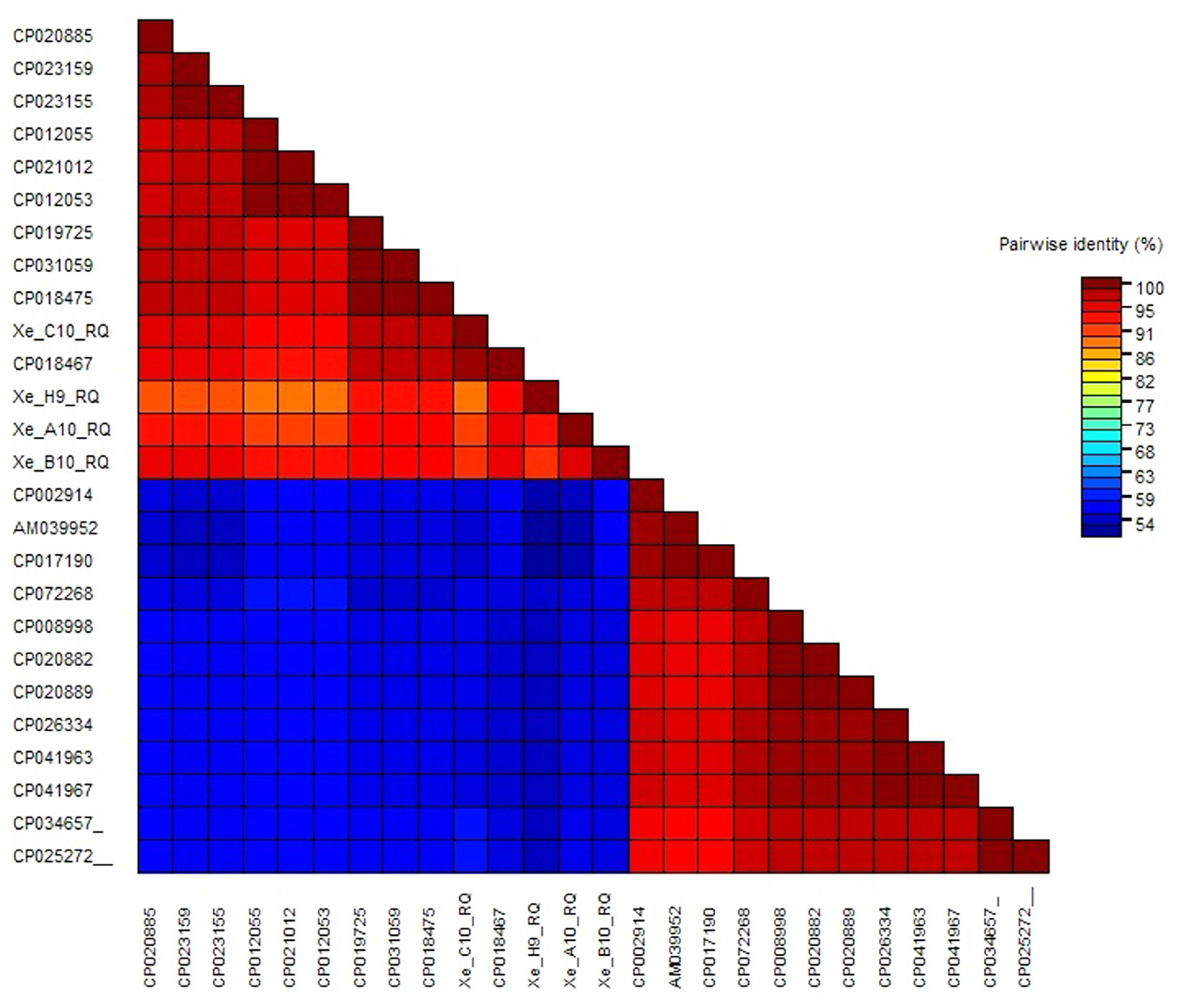
ANI calculation is one of the main aspects and approaches for comparative genomic data and taxonomic purposes. Furthermore, using OAT (Lee et al., 2015), ANI analysis revealed that pairwise identity between X. euvesicatoria and X. compestris pv. vesicatoria was 99.9%, indicating high similarity among the genomes, while X. axonopodis pv. citrumelo and X. perforans showed 98.48% and 98.46% similarity, respectively (Fig. 2). X. gardneri and X. vesicatoria found 86% ANI homology that was less similar as compared to other BS-causing Xanthomonas species. Additionally, X. euvesicatoria and X. compestris pv. vesicatoria, X. perforans, and X. axonopodis pv. citrumelo were clustered in one group and showed 99% to 98% homology, but X. gardneri and X. vesicatoria were recognized in another group and showed less homology.
Gene selection was aided by mauve-based progressive multiple genome sequence alignments. Genomes of X. campestris pv. vesicatoria (CP017190), X. perforans (CP019725), X. euvesicatoria (CP018467), X. axonopodis pv. citrumelo F1 (CP002914), X. gardneri (CP018731), and X. campestris pv. vesicatoria (AM039952) were aligned through progressive mauve software. Progressive mauve generates multiple genome alignments, which can be used for comparative genomic and population genomic investigations. In this study, multiple alignments of six genomes were used for the identification and location of ATP-dependent DNA helicase recQ gene (Supplemenary Fig. 1). In multiple alignments, each genome is laid out horizontally, with homologous portions indicated as colored rectangles in locally collinear blocks (LCBs). Syntenic regions showed by boxes of the same color, inverted regions engaged by boxes below the horizontal strain line, and rearrangement indicated by colored lines (Supplementary Fig. 1).
Multiple alignments showed that X. euvesicatoria and X. perforans were closely relative at regions inverted with homologous segments LCBs. On the other hand, X. axonopodis pv. citrumelo (CP002914), X. campestris pv. vesicatoria (CP017190) and X. campestris pv. vesicatoria (AM039952) were closely relative at syntenic regions. In contrast, X. gardneri (CP018731), was not much closer to X. euvesicatoria and X. perforans, some region was at inverted and some in the syntenic region. Lines collate aligned segments between genomes.
The gene encoding ATP-dependent DNA helicase recQ was located at position 3516837-3518633 bp in the genomes of X. euvesicatoria (CP018467), X. axonopodis pv. citrumelo F1 (CP002914), X. gardneri (CP018731), X. perforans (CP019725), X. campestris pv. vesicatoria (CP017190), and X. campestris pv. vesicatoria (AM039952) (Supplementary Fig. 1). Interestingly, the genes located upstream and downstream were highly similar in the six genomes. recQ gene was exploited as a target gene to identify X. euvesicatoria on the P. pubescens host and design primers for molecular techniques such as LAMP, PCR, and reverse transcription polymerase chain reaction (RT-PCR).
Homologous gene analysis of target genes recQ, hrpB1, and hrpB2
Comparative analysis of 13 genomes were also performed through GeConT (Ciria et al., 2004) using BLAST modules for specific genes localization and homologous gene analysis in the selected genomes. In this study, results showed that all the genomes showed recQ gene with different motifs in Figure. recQ gene motifs showed on the different protein families such as RecQ Zn binding, RQC domain, HRDC domain, DEAD/DEAH box helicase, helicase conserved C-terminal domain and protein of unknown function (DUF2385). Furthermore, recQ gene existing in superfamily II DNA helicase on the cluster of orthologous groups. recQ gene of X. campestris pv. vesicatoria 85-10 neighborhood to dpsA gene that is DNA-binding related protein (Fig. 3B). On the other hand, visualizing the genome context of the hrpB1 and hrpB2 genes was also analyzed. In all genomes, hrpB1 was shown on HrpB1_HrpK motif which indicated the bacterial type III secretion protein while hrpB2 showed on HrpB2 motif which also indicated the bacterial type III secretion protein. hrpB1 and hrpB2 genes also showed neighborhood genes (Fig. 3A).
PCR assay and confirmation of primers
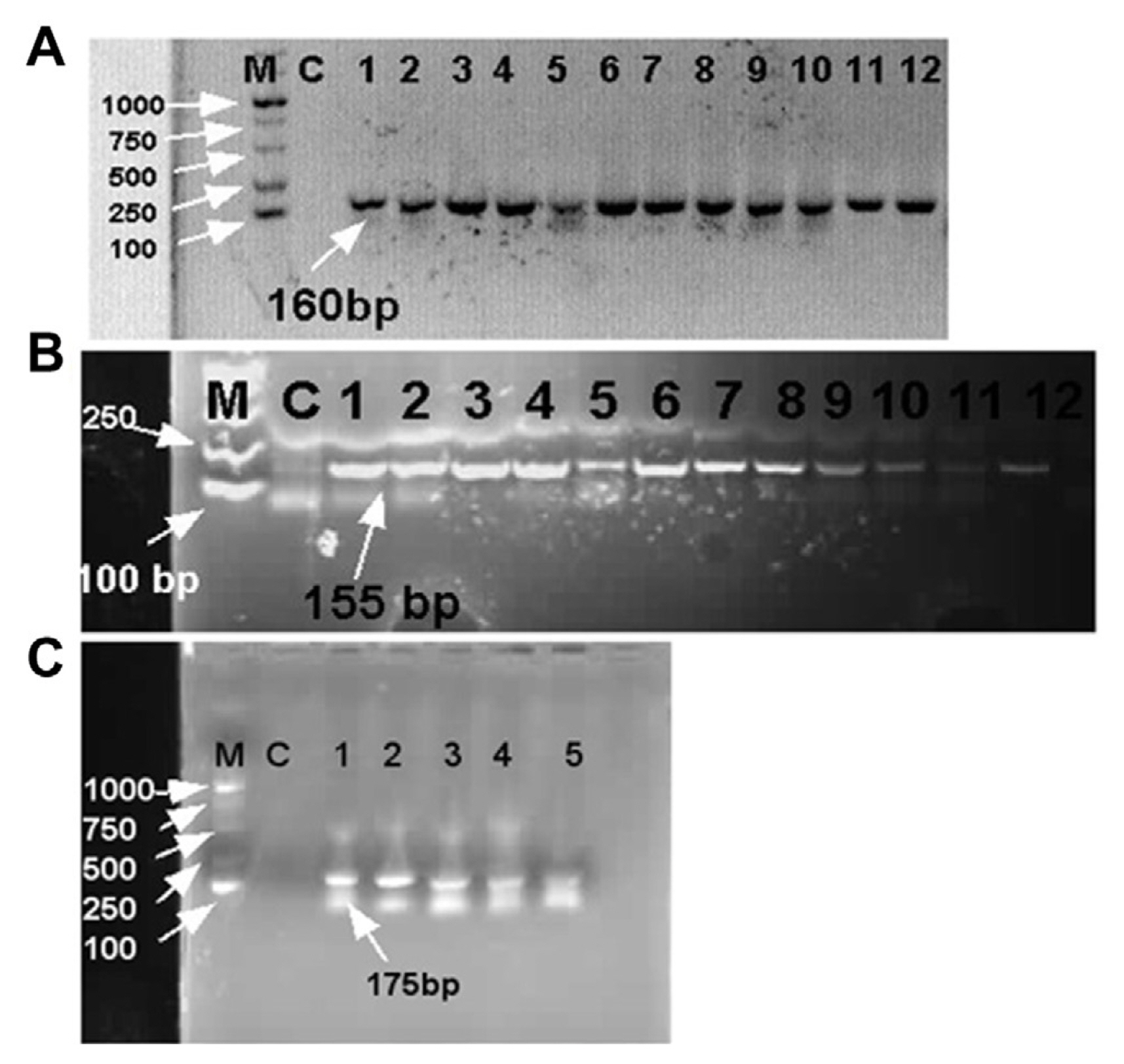
All designed primers were tested by Blastn in the NCBI GenBank database for confirmation of Xanthomonas spp., which revealed 100% query and identity with Xanthomonas species that cause BLS disease. Infected samples taken from the field were subjected to conventional PCR with hrpB1, hrpB2, and recQ genes. All of the infected samples showed positive amplification with amplicon size 160 bp, 155 bp, and 175 bp on 1% agarose gel electrophoresis using hrpB1, hrpB2, and recQ genes, respectively (Fig. 4). Therefore, positive amplification detected infected samples, whereas positive amplification was not observed in the negative control or healthy leaf samples. Furthermore, sequencing products were sent to Sangon Biotech (Shanghai) Co., Ltd. of three selective genes for molecular characterization and further analysis.
Efficiency of the LAMP assay
We used the LAMP assay with a variety of LAMP reagent doses, intervals, and temperatures to find the best reaction system for detecting X. euvesicatoria using the recQ ATP-dependent DNA helicase gene. Results indicate that the best concentrations of LAMP reaction for X. euvesicatoria were found at 1.6 ╬╝M for internal primers (FIP and BIP) followed by 0.2 ╬╝M of outer primers (F3 and B3) with ratio (1:8) and 0.8 ╬╝M for loop primers (LF and LB), 1.4 mM for dNTPs, 8 mM for MgSO4, 1├Ś isothermal amplification buffer, 1 U of Bst2.0 WarmStart DNA polymerase (Supplementary Fig. 2A-E). Optimum temperatures of 61┬░C, 63┬░C, and 65┬░C were observed and showed clear band on agarose gel (Supplementary Fig. 2B). The best temperature and time were found at 63┬░C for 45 min, and final melting temperature was conducted at 83┬░C for 10 min, and at the end of reaction 125 ╬╝M of HNB was used for color visualization. All positive samples of LAMP products were visualized amplification look like ladder-shaped bands and additionally, HNB dye analysis indicated that color change from violet to sky blue for positive amplification, whereas negative samples showed no change in color (Supplementary Fig. 2G). On a 2% agarose gel blemished with one-liter ethidium bromide, the reaction yields were examined; ladder-shaped bands confirmed the efficacy of the LAMP primers.
LAMP assay for sensitivity
The LAMP reaction utilized template target DNA collected from diseased samples using the extraction procedures mentioned above. The sensitivity of the LAMP was measured by 10-fold serial dilution of infected DNA of P. pubescens. The sensitivity of the LAMP test ranged from 1 ├Ś 10ŌłÆ1 ng/╬╝l to 1 ├Ś 10ŌłÆ5 ng/╬╝l when observed with the naked eye. To confirm the amplification, the LAMP reaction products were run through a 2 percent agarose gel electrophoresis (Supplementary Fig. 2F). Sensitivity of template DNA for LAMP reaction ranged from 1 ├Ś 10ŌłÆ1 ng/╬╝l to 1 ├Ś 10ŌłÆ4 ng/╬╝l was observed but the amplification of 1 ├Ś 10ŌłÆ5 ng/╬╝l was not clearly shown on agarose gel electrophoresis.
LAMP and real-time PCR based assay for detection of infected field samples
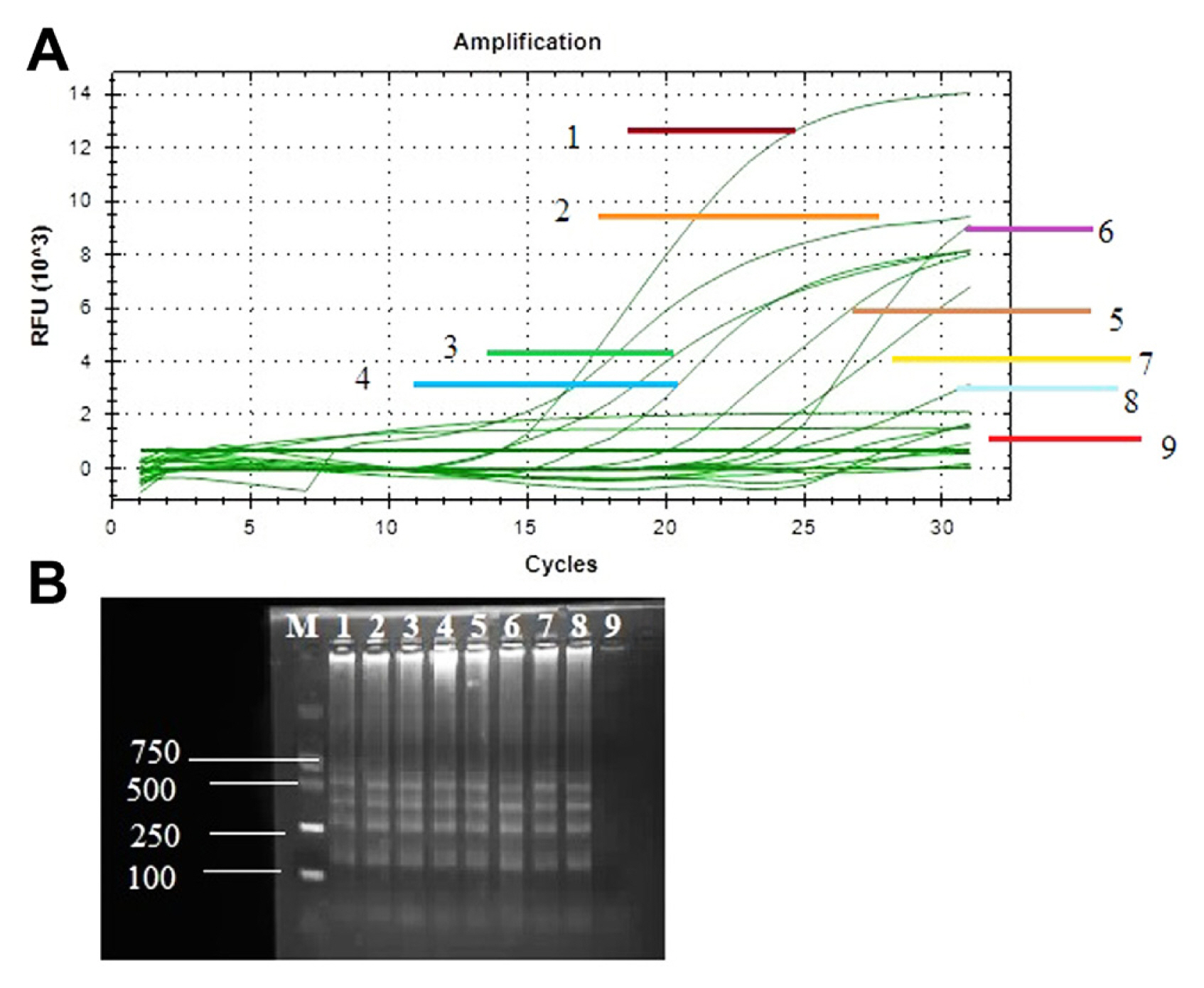
The disease samples were collected from different parts of the region of Heilongjiang Province, China. The performance of the LAMP assay and real-time PCR with specific target gene recQ, used for rapid and sensitive identification of all diseased samples. Positive amplification showed different ladder-shaped bands on 2% agarose gel electrophoresis while the negative sample showed no band on agarose gel electrophoresis. Therefore, infected samples showed positive amplification with amplified genes and no amplification product was obtained from NTC (Fig. 5B). Real-time PCR assay was also performed for sensitivity analysis of diseased samples. Amplification curves were observed for recQ genes in diseased and healthy samples. Positive amplification showed top curves from the bottom and reach their plateau between 7 relative fluorescence units (RFU) 103 to 14 RFU103 and negative amplification observed curve at the bottom of plot with zero RFU103 value (Fig. 5A).
Phylogenetic tree analysis and model design
Phylogenetic analysis is important for analyzing developmental events that occur during evolution, as well as gaining information on biological diversity and genetic classifications. It also depicts the evolutionary history or affiliation between different species, individuals, or organism characters that have adapted from a common ancestor.
In this study, sequence of hrpB1, hrpB2, and recQ genes were used for constructed the phylogenetic tree, and all the phylogenetic trees were conducted in MEGA 11 and furthermore, for well explained the phylogenetic trees designed by itol (https://itol.embl.de/). Model assortment is deliberated as an important phase in the process of building an evolutionary tree. We performed phylogeny inference under common models of DNA sequence evolution. Major 24 fit models of nucleotide substitutions designed in which assessed different models, i.e., T92, K2, HKY, TN93, JC, and GTR joined using the proportion of invariable sites (+I), rate heterogeneity across sites (+G), or both (+I+G) (Supplementary Table 1). Results showed that Tamura 3-parameter (T92) model was best the model for building evolutionary tree through ML method of nucleotide substitution. The Bayesian information criterion (BIC) (Schwarz, 1978) and corrected Akaike information criterion (Hurvich and Tsai, 1989) criterion were used to determine the goodness-of-fit of each model to the data. Tamura 3-parameter (T92) model was obtained the lowermost BIC scores as compared to another modal in Supplementary Table 1. Thus, in this study Tamura 3-parameter (T92) model was used as for more accurate evolution of building evolutionary tree by ML method.
Phylogenetic tree analysis with recQ, hrpB1, and hrpB2 genes
In this study, the evolutionary history of the sequences of recQ, hrpB1, and hrpB2 were inferred by using more prominent evolutionary history method the Neighbor-Joining method with bootstrap 1,000 replicates (Saitou and Nei, 1987). The evolutionary distances were computed using the Maximum Composite Likelihood method (Tamura et al., 2004). This analysis involved 48 nucleotide sequences. All ambiguous positions were removed for each sequence pair. There were a total of 759 positions in the final dataset. Evolutionary analyses were conducted in MEGA11 (Tamura et al., 2021). Sequences of different organisms used in this study were retrieved from NCBI database and all sequences were aligned through ClustalW. Evolutionary results showed that the sequence of strain XeHB1 was tightly clustered and homology corresponding with strain X. euvesicatoria LMG930 and in contrast, no strains were found to X. vesicatoria and X. gardneri (Fig. 6B). Furthermore, single gene phylogenetic tree analysis using hrpB1 gene was also performed for more accurate analysis. It was determined that the ML approach and the Tamura 3-parameter (T92) model were the best tools for deducing the evolutionary history (Tamura, 1992). Initial tree(s) for the heuristic search were obtained automatically by applying the Neighbor-Join and BioNJ algorithms to a matrix of pairwise distances estimated using the Tamura 3-parameter (T92) model, and then selecting the topology with the superior log likelihood value. The tree has been drawn to scale, and the length of each branch is indicated by the number of substitutions that have occurred at each site. In this particular investigation, there were 38 nucleotide sequences involved. The completed dataset contained 520 positions in total across its entirety. These results reveal that strain XeHB1 was 99-100% homology corresponding with strain X. euvesicatoria LMG930 and X. perforans LH3 (Fig. 6A). On the other hand, strain XeHR2 was closely relative to X. campestris pv. vesicatoria (strain 85-10) (Fig. 6B). Therefore, evolutionary tree analysis with hrpB1 and hrpB2 genes showed that all the reference strains were closely relative to X. euvesicatoria, X. campestris pv. vesicatoria (strain 85-10) and did not find X. vesicatoria and X. gardneri. The results of a whole genome comparison between X. euvesicatoria LMG930 and X. campestris pv. vesicatoria (strain 85-10) revealed that the sequences of both genomes were closely related, with an ANI of 99.94% (Fig. 2). The evolution of the recQ gene demonstrated that the strain XeB10RQ was more compatible with the strain X. euvesicatoria LMG930 (Fig. 6B).
Phylogenetic tree analysis with specific target gene recQ helicase
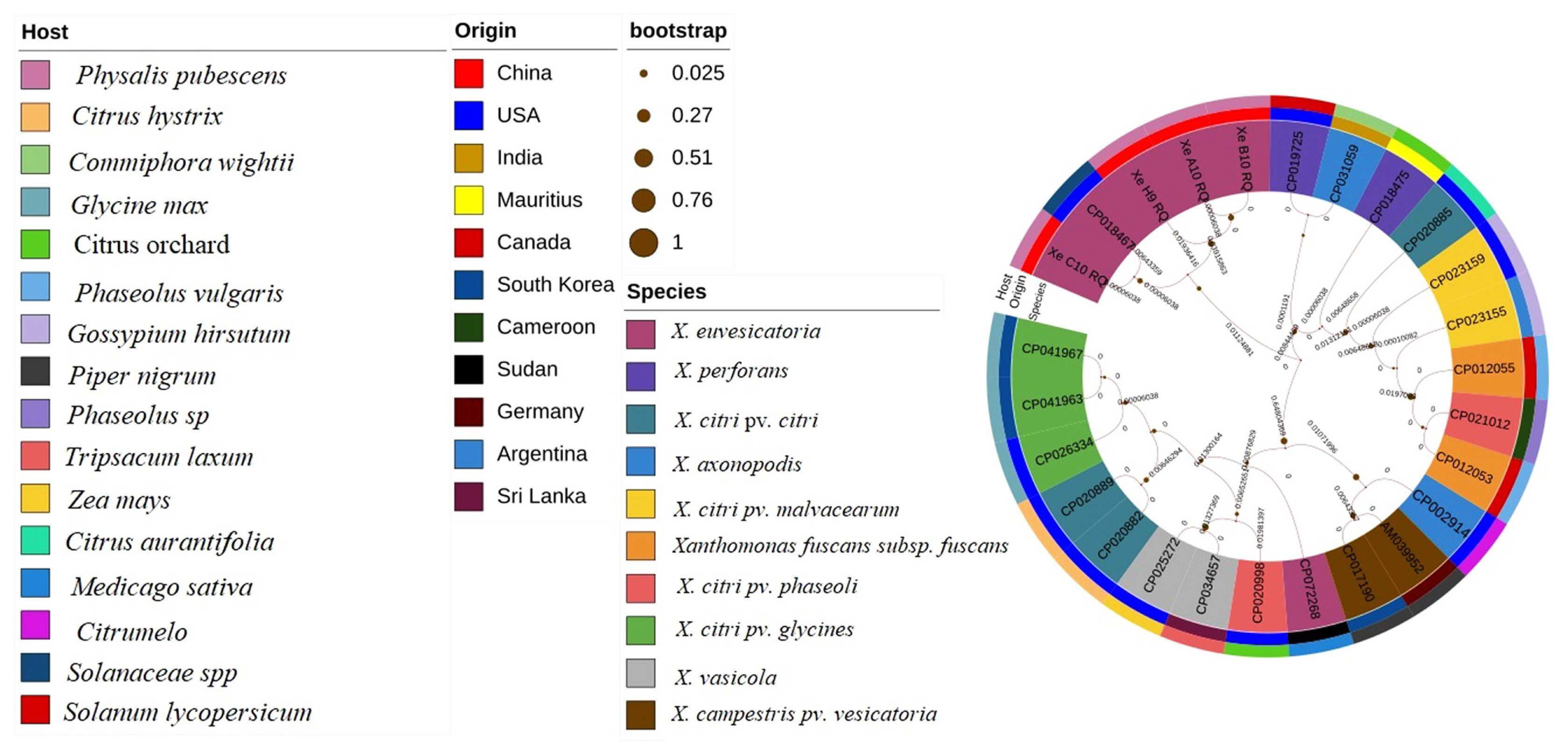
Evaluation of strains X. euvesicatoria with specific target gene recQ was conducted through evolutionary history analysis. Comparative analysis of different sequences from target gene recQ were retrieve from NCBI database with specific host, origin and species. Phylogenetic tree analysis was conducted for the evaluation of the four strains XeC10RQ, XeH9RQ, XeA10RQ, and XeB10RQ with other Xanthomonas strains. Other species of Xanthomonas included X. citri pv. citri, X. fuscans subsp. fuscans, X. axonopodis pv. citrumelo, X. vasicola, X. vasicola pv. vasculorum, and X. citri pv. glycines that cause different diseases such as citrus canker, common bacterial blight, citrus bacterial spot, Xanthomonas wilt, bacterial leaf streak, and bacterial pustules, respectively. Phylogenetic tree analysis was inferred by most common method the ML method with 1,000 bootstraps replicates. Results showed that X. euvesicatoria strains XeC10RQ, XeH9RQ, XeA10RQ, and XeB10RQ which originated from China on the host P. pubescens were closely relative and homology with the strain X. euvesicatoria LMG 930 (accession no. CP018467) which origin from USA on Piper nigrum (Fig. 7). Evolutionary history showed that these four strains were no found homology with X. perforans, X. axonopodis, X. vesicatoria, and X. gardneri that cause BLS disease.
Furthermore, color-coded matrix analysis was designed for pairwise identical sequences. Color-coded matrix results showed that the sequence of the strain XeC10RQ was found 100% homology and identical of the sequence with the strain X. euvesicatoria LMG 930 (CP018467) in contrast other three strains showed 95% to 98% homology with the strain X. euvesicatoria LMG 930 (CP018467) (Fig. 8). All Xanthomonas strains details with accession number, host, origin disease and reference existed in Supplementary Table 2.
Pathogenicity, confirmation, and disease severity analysis of X. euvesicatoria using hrpB1, hrpB2, and recQ
All inoculated leaves of P. pubescens showed Xanthomonas signs of bacterial spot. Symptoms were observed after 3, 7, 14, and 21 days of post-inoculation. Infection was characterized by water-soaked lesion, rupture leaf lamina, necrotic lesion with a yellow margin, necrosis with chlorosis margin, and deformation of leaf but in some leaves showed a black spot on lesion, and after that turn to black color with greasy appearance, whereas bacterial population grows on nutrient agar showed yellow colonies and healthy leaves used as negative control (Supplementary Fig. 3).
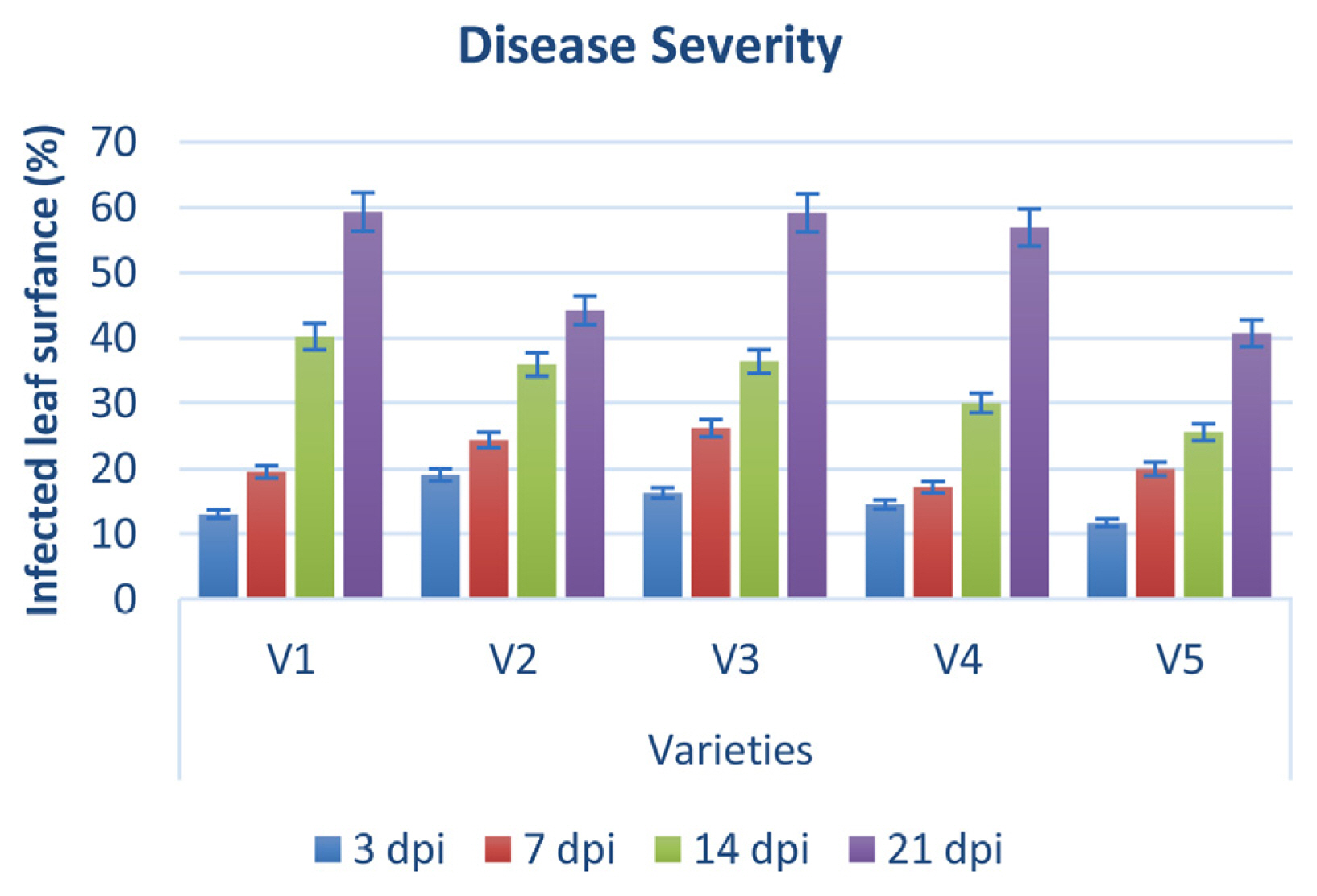
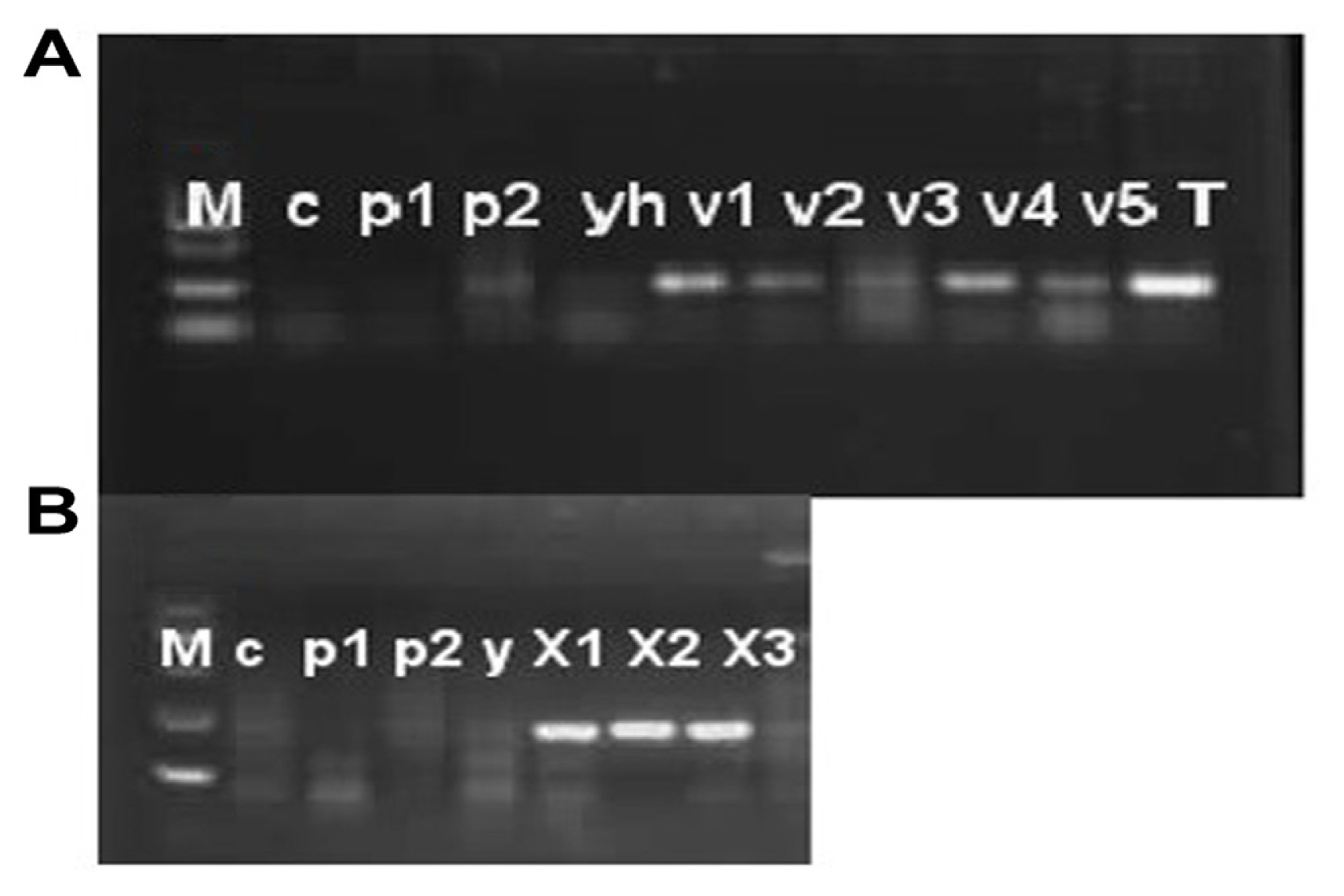
Disease severity was observed on five cultivars of P. pubescens and measured by Canopeo app (Patrignani and Ochsner, 2015) after 3, 7, 14, and 21 days of post-inoculation. Bar graph results showed that disease severity is separated into three typesŌĆÖ low severity, intermediate severity, and high severity. Three and 7 days post inoculation existed into low severity and 14 days post inoculation was occurrence in medium severity, while 21 days post inoculation showed high severity (Fig. 9). Low severity was observed between percentage ranges from 13% to 25%, while high severity was observed from 40% to 59% of infected damage surface after inoculation (Fig. 9). PCR analysis was performed for confirmation of X. euvesicatoria and specificity of hrpB1, hrpB2, and recQ gene primers. Fifty-two bacterial strains (Supplementary Table 3) were used for the evaluation of tested primers specificity and PCR results showed positive amplification through recQ, hrpB1 and hrpB2 genes (Supplementary Fig. 4). Furthermore, type bacterial strains of X. euvesicatoria, X. vesicatoria, X. gardneri, and X. perforans were obtained from Northeast Agricultural University, Harbin, China. Inoculated samples (v1, v2, v3, v4, v5, and T) were used as reference strains while other bacterial strains (P. syringae pv. syringae and X. oryzae pv. oryzae) used for specificity of primers and healthy leaves for negative control. Inoculated samples (v1, v2, v3, v4, v5, and T) indicated positive amplification, whereas two strains of P. syringae pv. syringae (Pss1 and Pss2), X. oryzae pv. oryzae strain (YH) and healthy leaf (c) showed negative amplification on 1% agarose gel electrophoresis (Fig. 10A). Furthermore, PCR analysis was conducted for confirmation of X. euvesicatoria with hrpB1, hrpB2, and recQ genes by using strains Xeu1, Xeu2, and Xeu3 that were collected from Song et al. (2019) (Fig. 10B).
Discussion
P. pubescens is mostly grown in Heilongjiang Province, ChinaŌĆÖs northeast and there are numerous additional informal Chinese names, such as ŌĆ£gu niaoŌĆØ and ŌĆ£mao suan jiang.ŌĆØ Its fruit is a sphere-shaped berry that ripens from green to yellow and has a diameter of 1.25 to 2.50 cm. It is fully coated with a flimsy tenacious calyx through its development and ripening. (Luchese et al., 2015). BLS is a worldwide problem caused by four Xanthomonas species such as X. euvesicatoria, X. gardneri, X. perforans, and X. vesicatoria. Under unfavorable conditions, these four species can cause a 50% yield loss in tomato and pepper plants (Dhakal et al., 2019) but now a day, first identified X. euvesicatoria pv. euvesicatoria on P. pubescens and caused severe economic losses reported by Song et al. (2019). In our study, 52 pathogenic isolates were obtained from bacterial spot lesions on P. pubescens plants throughout Heilongjiang Province, China.
Comparative genome and phylogenetic tree analysis have important factors to illustrate the commonalities and dissimilarities in structure and function (Alzahrani et al., 2021). Next-generation sequencing technologies have established a framework for searching for unique gene sequences, permitting the development of highly specific, trustworthy, and robust field-deployable assays (Ouyang et al., 2013). In our study, we performed a comparative genome analysis of 13 genomes that are closely relative to each other through two more prominent methods Blast comparison and ANI, and data were retrieved from NCBI database facilitating identification and selection of specific target gene ŌĆ£recQŌĆØ. recQ helicases are important components of DNA renovation and recombination processes involved in genome integrity (Hartung and Puchta, 2006). Following an evolutionary approach, we explored the DNA repair gene in the genomes of the Xanthomonadales group to better understand the methods by which these organisms respond to environmental stressors, such as plant infection. Our comparative genome results showed that the whole genome sequence of X. euvesicatoria strain LMG930 was closely relative and 98-99% homology with genome sequence of X. compestris pv. vesicatoria strain 85-10 X. perforans LH3 and X. axonopodis pv. citrumelo F1 (Figs. 1 and 2). In contrast, X. euvesicatoria strain LMG930 found 93%, 89%, 86%, 86%, 85%, 85%, 85%, and 78% homology with X. citri pv. glycines, X. vasicola, X. hortorum pv. gardneri, X. arboricola, X. fragariea, X. vesicatoria, X. cucurbitea, and X. translucens, respectively. Therefore, phylogenetic analysis of whole genome sequence through ANI showed clearly that strain X. euvesicatoria is very closely relative to strains X. compestris pv. vesicatoria strain 85-10 X. perforans LH3 and X. axonopodis pv. citrumelo F1 while other strains were found large genetic diversity with X. euvesicatoria.
recG, a gene specific to X. euvesicatoria, was discovered primarily to the employment of MAUVE to investigate the evolution of Xanthomonas species on a large scale (Larrea-Sarmiento et al., 2018). Darling et al. (2010) emphasized that the multiple genome alignments generated by MAUVE progressive for comparative genomic, population genomic studies, and selection and location of genes in the genomes. In our study, using MAUVE multiple alignments to find out location of target gene, we emphasized that the recQ gene, which encodes the ATP-dependent DNA helicase, was found in the genomes of X. euvesicatoria, X. axonopodis pv. citrumelo F1, X. gardneri, X. perforans, and X. campestris pv. vesicatoria at the position of 3,516,837-3,518,633 bp in all genomes.
In previous studies, molecular diagnosisŌĆÖs potential for X. euvesicatoria through multiplex PCR, nested-PCR, real-time PCR (qPCR), conventional PCR, LAMP-PCR, droplet digital PCR, fluorescence in situ hybridization, and Box element PCR (BOX-PCR) have been described (Kositcharoenkul et al., 2011; Munhoz et al., 2011). PCR based techniques have been used for pathogen recognition for more than 30 years and are one of the numerous DNA-based fast approaches. In contrast, different researchers (Larrea-Sarmiento et al., 2018; Strayer et al., 2016; Yasuhara-Bell et al., 2017) focus on LAMP technology as compared to PCR. However, we compared conventional PCR, LAMP, and RT-PCR were used for rapid, accurate, and more sensitive identification of X. euvesicatoria on P. pubescens. Concluded that among molecular approaches LAMP was less timely and costly for pathogen diagnosis detection rather than PCR and qPCR analysis. Molecular evolution of X. euvesicatoria on P. pubescens through conventional PCR with selected genes hrpB1 and hrpB2 type III-secreted proteins that are mostly critical for pathogenicity and detection of Xanthomonas pathogens. According to Teper et al. (2016), a T3SS pathway transports effector proteins into host cells to overcome plant resistance and encourage disease influences X. euvesicatoria pathogenicity.
Our PCR results showed that all the infected samples were found positive amplification with 160 bp and 155 bp, respectively on agarose gel electrophoresis (Fig. 4). In contrast, we also developed validated and a colorimetric-based LAMP approach for detecting X. euvesicatoria on P. pubescens that is specific, sensitive, reliable, and resilient. LAMP optimization analysis revealed that concentrations of primer, Bst and MgSO4 were found at 1:8, 1, and 8 mM, respectively on the 61┬░C at 45 min. Moreover, in our study, betaine concentrations (0.4, 0.6, 0.8, 1, 1.2, and 1.4 M) did not find significantly affected by chemicals reaction. Positive results showed clearly like as different ladder band on agarose gel electrophoresis.
Furthermore, for more accurate results colorimeter analysis developed through HNB and positive amplifications were found the color change from dark blue to violet sky and negative samples showed no change in color. BLS four infections generated considerable economic losses, according to Larrea-Sarmiento et al. (2018) and they also compared the genomes of X. gardneri, X. vesicatoria, X. euvesicatoria, and X. perforans to those of other Xanthomonas species, and the gene recG was utilized to construct primers for a LAMP assay for fast and effective identification. We compared all molecular approaches for investigation and quick detection of X. euvesicatoria and concluded that LAMP has a novel technique for addressing the shortcomings of PCR-based technologies (Niessen, 2015). The most widely used isothermal-based detection technology is LAMP due to its rapidity, ease of use, increased sensitivity, and compatibility with a variety of detection chemicals. Most importantly, it is simple to do at the point of care as compared to PCR-based detection methods.
The sensitivity of the fabrication LAMP test was verified to ratify the finding bounds in the presence and absence of tested DNA. Sensitivity of LAMP fluctuates by the pathogen, according to Schrader et al. (2012), possibly related to bacterial functional features such as extracellular polysaccharide-producing vs. non-producing bacteria. We observed the sensitivity of the LAMP was tested using a 10-fold serial dilution of P. pubescens infected DNA ranging from 1 ├Ś 10ŌłÆ1 to 10ŌłÆ5 ng/╬╝l. The sensitivity of template DNA for the LAMP reaction ranged from 1 ├Ś 10ŌłÆ1 to 10ŌłÆ4 ng/╬╝l because amplification of 1 ├Ś 10ŌłÆ5 ng/╬╝l on gel electrophoresis was not clearly visible.
Among evolutionary methods, phylogenetic tree is more reliable to find out evolutionary history and detection of X. euvesicatoria with specific target or housekeeping genes (Song et al., 2019). Many researchers have studied the detection of X. euvesicatoria through the phylogenetic trees on tomato and pepper plants (Kyeon et al., 2016; Roach et al., 2018; Yaripour et al., 2017). In this study, phylogenetic tree analysis was performed with T3SS genes (hrpB1 and hrpB2) with 1000 bootstraps through ML method and found that strains XeHB1 and XeHR2 were closely relative to X. euvesicatoria strains (LMG930), X. perforans strains (LH3), and X. campestris pv. vesicatoria (strain 85-10) and this indicates that all tested strains existed in X. euvesicatoria because ANI and blast comparison results indicated that the genome of X. euvesicatoria showed 99.94% and 98% ANI with X. perforans and X. campestris pv. vesicatoria (strain 85-10) respectively whereas, no tested strains were found homology with X. vesicatoria and X. gardneri. A circular phylogenetic tree using recQ gene showed that four strains XeC10RQ, XeH9RQ, XeA10RQ, and XeB10RQ isolated from P. pubescens that collected in Heilongjiang Province, China were very closely relative and maximum homology with strain X. euvesicatoria LMG930 from USA on Piper nigrum. Furthermore, color-coded pairwise identity matrix showed that XeC10RQ, XeH9RQ, XeA10RQ, and XeB10RQ found 95-99% pairwise identity with strain X. euvesicatoria because recQ has the ability to DNA repair and recombination pathways elaborate in the looking after of genome reliability (Hartung and Puchta, 2006).
Pathogenicity studies revealed that X. euvesicatoria was found on all inoculated P. pubescens as well as tomato plants (Jones et al., 2004). Following 3, 7, 14, and 21 days after inoculation, symptoms were detected (Supplemenary Fig. 3). The Canopeo app measured disease severity 3, 7, 14, and 21 days after inoculation (Patrignani and Ochsner, 2015). The severity of the disease was categorized into three types: low, intermediate, and high. After inoculation, low severity was observed between percentage ranges of 13% to 25%, whereas high severity was reported between 40% and 59% of infected damage surface (Fig. 9). According to Hern├Īndez-Huerta et al. (2021), Xanthomonads were grouped into three groups in a cluster analysis of bacterial spot severity according to their virulence and findings that isolated X. euvesicatoria caused medium damage 76.8% final severity and low damage 29.8% final severity.


 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement1
Supplement1 Print
Print