Rice is a major staple food for over a half of the worldŌĆÖs population in charge of more than 20% of their daily calorie intake (Seck et al., 2012; Skamnioti and Gurr, 2009). Its stable production is socioeconomically important in international food security. Rice blast disease caused by the ascomycete Magnaporthe oryzae occurs in every ricegrowing region worldwide, resulting in serious damage to rice production (Kato, 2001; Ou, 1985). High variability of this fungal population often leads to break down of resistance in newly-released rice cultivars in the fields because new races which overcome the resistance are frequently emerged out of the mixed population of M. oryzae (Ou, 1980; Valent and Chumley, 1991; Zhu et al., 2000). Understanding the structure and the dynamics of the pathogen population is essential for development of sustainable strategies for disease management of rice blast (Meung et al., 2003).
One of the popular markers for characterizing population of M. oryzae is restriction fragment-length polymorphism (RFLP) analysis using repetitive DNA sequences called MGR (Magnaporthe grisea repeat) (Borromeo et al., 1993; Hamer et al., 1989). Since MGRs are characteristically abundant in the genome of the rice-infecting isolates (Dean et al., 2005), the numbers of bands in Southern hybridization assay can distinguish between the rice-infecting and the weed-infecting isolates (Hamer et al., 1989). Furthermore, MGR-RFLP has been employed to characterize population dynamics of rice-infecting isolates in different populations over the world (Levy et al., 1991; Levy et al., 1993; Park et al., 2003; Xia et al., 1993; Zeigler et al., 1995). Although MGR-RFLP has limitations in correlating genotypes with pathotypes within the rice-infecting populations, it is still a useful marker in understanding the genetic diversity of fungal populations. However, MGR-RFLP is time-consuming, labor-intensive and is not based on one-locus alleles which are used in measuring classical population parameters (Adreit et al., 2007; Brondani et al., 2000).
Microsatellites, also known as Simple Sequence Repeats (SSR), are tandemly repeated sequences of short length DNA (usually 2-6 bp) found in all prokaryotic and eukaryotic genomes (Zane et al., 2002). They show variation in the number of repeats at a specific locus and strand slippage replication is generally considered to contribute to such variation (Levinson and Gutman, 1987; Tautz, 1989). Due to their prevalence and abundance in the genome, easy detection by PCR, and length polymorphism at a given locus, they have become a popular and powerful molecular marker for genetic and physical mapping, forensic DNA studies, and population genetics (Jarne and Lagoda, 1996; Tautz, 1989).
Since the genome sequence information of M. oryzae became publicly available (Dean et al., 2005), the computational prediction and selection of these markers was made possible (Karaoglu et al., 2005; Lim et al., 2004). A total of 685 of polymorphic microsatellite markers have been developed by several research groups to analyze population structure and construct genetic map (Adreit et al., 2007; Brondani et al., 2000; Feng et al., 2007; Kaye et al., 2003; Suzuki et al., 2009; Zheng et al., 2008). However, most of the markers have only been used for genotyping fewer than ten rice isolates or a progeny population generated from a cross. For example, 120 and 313 microsatellite markers were developed by in silico prediction of the M. oryzae genome sequence but tested for only three and nine isolates, respectively (Feng et al., 2007; Zheng et al., 2008). Only a small exception of 30 markers were applied to field population with more than 50 isolates (Adreit et al., 2007; Brondani et al., 2000; Suzuki et al., 2009) among which 18 markers were used to characterize the populations (173 to 645 isolates) of M. oryzae collected from Africa, Asia, and Europe (Adreit et al., 2007). Here, we used microsatellite markers to analyze the structure and dynamics of the Korean population collected over two decades (Park et al., 2003). In addition, fluorescence-labeled primers and automated DNA sequencing were used in digitization of allele information which allows a global comparative analysis (Oda et al., 1997; Toh et al., 1996).
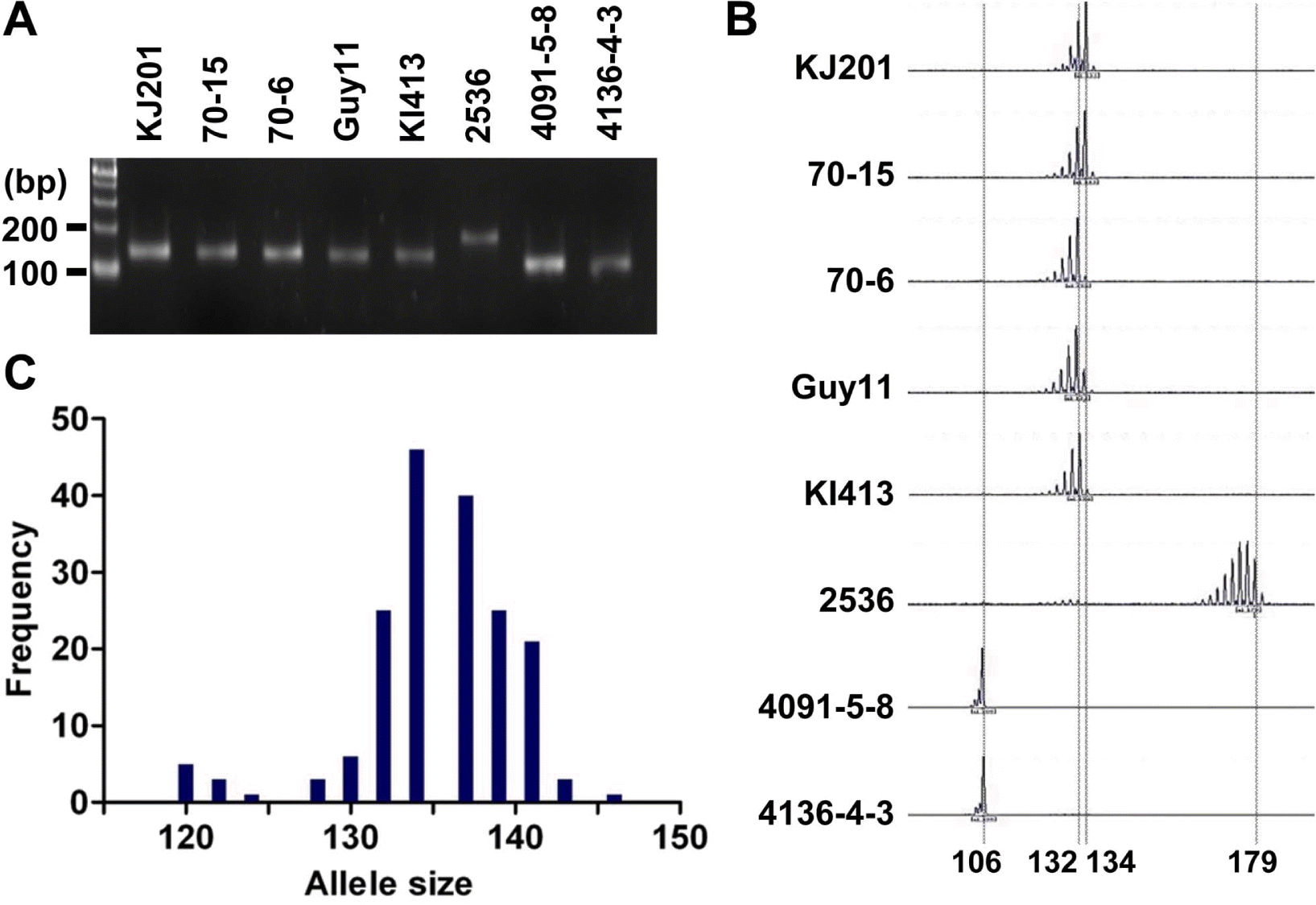
As a part of the global collaboration for universal marker development, information of 25 microsatellite markers was generously provided by Dr. D. Tharreau (CIRAD, France). Initially, the markers were tested in eight representative M. oryzae strains: Guy11, KJ201, KI413, 70-15, 70-6, 2539, 4091-5-8, and 4136-4-3 (Guy11 is a hermaphroditic rice pathogen isolated from the French Guiana (Leung et al., 1988); KJ201 and KI413 are rice-infecting isolates from Korea; 70-15 and 70-6 are laboratory strains from numerous back crosses to Guy11 (Chao and Ellingboe, 1991); in addition, 70-15 is a reference strain used for whole genome sequencing (Dean et al., 2005); 4091-5-8 and 4136-4-3 originated from a cross between a weeping lovegrass pathogen (K76-79) and a finger millet and goosegrass pathogen (WGG-FA40) (Valent et al., 1991); 2539 is originated from a cross between fertile weed isolates (Leung et al., 1988)). PCR was performed in a GeneAmp PCR system 9700 (Applied Biosystems, CA, USA) using 10 to 30 ng of genomic DNA as a template. Cycling conditions were 95 ┬░C for 5 min, then 30 cycles of 95 ┬░C for 10 sec, 55 ┬░C for 30 sec, 72 ┬░C for 30 sec, and a final extension at 72 ┬░C for 5 min. PCR products were analyzed on 3% agarose gels (mixture of 1.5% agarose and 1.5% low-melting agarose) to determine length variation (Figure 1A). If no product was found in one of the eight strains or all strains showed different sizes of PCR product, the marker was discarded (data not shown). Finally, 11 microsatellite markers were selected for further genotyping analysis of the Korean population of M. oryzae. The Korean population had been collected from different spots in Korea for two decades since the 1980ŌĆÖs (Park et al., 2003). Genomic DNAs of the 190 isolates were obtained from the Center for Fungal Genetic Resources (Seoul National University, Korea; http://genebank.snu.ac.kr). For high resolution genotyping using microsatellite markers, one of the primers for each microsatellite locus was labeled with a fluorescent dye (6-FAM, PET, VIC, or NED). After PCR reactions using the fluorescent primers, 5 ╬╝l of PCR products labeled with different fluorescent dyes were pooled to make 20 ╬╝l of mixture for multiplex assays. Then, 0.5 ╬╝l of the mixture was added to 9 ╬╝l of formamide and 0.5 ╬╝l of the GeneScan 500 LIZ size standard (Applied Biosystems, CA, USA). The final mixtures (10 ╬╝l) were loaded on a 3100 Genetic Analyzer (Applied Biosystems, CA, USA) after denaturation. Allele sizes were digitized using GeneMapper software version 3.7 (Applied Biosystems, CA, USA) (Figure 1B).
The number of alleles detected per locus ranged from 2 to 17 with an average of 6.5 (Table 1). Gene diversity (HE = 1 ŌłÆ ╬Żxi2, where xi is the frequency of the ith allele) was calculated using the GENETIX 4.05.2 software (Belkhir et al., 1996ŌłÆ2004). For all the populations examined, HE ranged from 0.021 to 0.838 with an average value of 0.412 (Table 1). No null allele was found at the loci in this study. Comparative analysis was performed with the gene diversity data of the Korean population and six previously reported populations in the world (Adreit et al., 2007). The data for eight markers commonly used in both studies were compared in further analyses. In allele diversity and HE, the Korean population exhibited significant positive correlation (r Ōēź 0.844) with the populations of Spain and France (P < 0.01). Remarkably, the range of allele diversity in Korea (2 to 17) is similar to the integrated diversity of the six populations (4 to 19) showed that the Korean population is highly diverse (Figure 2). DNA fingerprinting analysis also supported high genetic diversity of the Korean population and no clear lineage was supported by a high bootstrap value (Park et al., 2003). To examine how loci are associated with each other, genotypic linkage disequilibrium (LD) was tested across all pairs of loci in the population using GENEPOP 4.0.10 (Raymond and Rousset, 1995). Contingency tables were generated for all pairs of loci in the population. A G test (log-likelihood ratio) was performed for all tables using the Markov chain algorithm based on the null hypothesis that genotypes at one locus are independent of those at the other locus. Significant LD was found in 33 out of 55 pairs of loci (P < 0.05), suggesting that the population tested might mainly depend on asexual reproduction.
In this study, comparative analysis for allele frequency using multilocus microsatellite typing with 11 markers revealed that the Korean population is highly diverse because its allele frequency was similar to the integrated frequency of six geographic populations. These microsatellite markers and comprehensive collection of the Korean isolates are useful resources for studying genetic diversity of M. oryzae populations and understanding their evolution history.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print