Development of the Droplet Digital PCR Method for the Detection and Quantification of Erwinia pyrifoliae
Article information

Abstract
Black shoot blight disease caused by Erwinia pyrifoliae has serious impacts on quality and yield in pear production in Korea; therefore, rapid and accurate methods for its detection are needed. However, traditional detection methods require a great deal of time and fail to achieve absolute quantification. In the present study, we developed a droplet digital polymerase chain reaction (ddPCR) method for the detection and absolute quantification of E. pyrifoliae using a pair of species-specific primers. The detection range was 103–107 copies/ml (DNA templates) and cfu/ml (cell culture templates). This new method exhibited good linearity and repeatability and was validated by absolute quantification of E. pyrifoliae DNA copies from samples of artificially inoculated immature pear fruits. Here, we present the first study of ddPCR assay for the detection and quantification of E. pyrifoliae. This method has potential applications in epidemiology and for the early prediction of black shoot blight outbreaks.
Asian pear (Pyrus pyrifolia) black shoot blight caused by Erwinia pyrifoliae is among the most destructive agricultural diseases in South Korea (Shrestha et al., 2003). Symptoms observed on infected trees include dark-brown stalks and flowers and immature fruit dropping, very similar to the symptoms of fire blight in apple and pear caused by Erwinia amylovora (Shrestha et al., 2003). Since first reported in 1999, E. pyrifoliae has been responsible for severe losses in fruit production in South Korea. Moreover, E. pyrifoliae can also infect apple trees, manifesting as brownish blight (Han et al., 2016). Despite efforts by the Korean government to manage and control the disease, including the eradication of infected orchards and burial of diseased plants, this pathogen was isolated in consecutive years and sporadic outbreaks continue to occur in South Korea (Geider et al., 2009; Kim et al., 1999; Rhim et al., 1999). Recently, E. pyrifoliae has also been reported as a newly discovered pathogen on strawberry in the Netherlands (Wenneker and Bergsma-Vlami, 2015). Due to the economic importance of apple and pear production and the reduction in fruit production and quality caused by E. pyrifoliae (Kim et al., 2001; Shrestha et al., 2003), a reliable, rapid, and accurate method for the identification and early detection of this pathogen is required.
Plant pathogens are generally identified using culture-based methods, including pathogen isolation, incubation, and pathogenicity testing to fulfill Koch’s postulate; these approaches are time consuming and laborious (Du et al., 2021). With the development of molecular technologies, diverse DNA-based methods such as conventional polymerase chain reaction (PCR), quantitative real-time PCR (qPCR), and loop-mediated isothermal amplification (LAMP) have been used for non–culture-based identification and detection of plant pathogens. However, conventional PCR has some limitations, including its low specificity and relatively high detection threshold, and the inability to quantify precisely copy numbers of target genes. qPCR methods have been adopted widely for pathogen detection and quantification in various fields because of their specificity and sensitivity (Chai et al., 2020; Dinu and Bach, 2013; Luo et al., 2008; Telli and Doğruer, 2019). In addition, the LAMP PCR assay can be performed at a constant temperature within a short reaction time, presenting a rapid and simple method to detect specific pathogens for on-site diagnosis (Shin et al., 2018; Yamazaki et al., 2008; Zhang et al., 2018).
Droplet digital polymerase chain reaction (ddPCR), a third-generation PCR technology, has emerged as an alternative to qPCR and other quantitative methods (Huggett et al., 2016; Lei et al., 2020; Li et al., 2018; Voegel and Nelson, 2018). The ddPCR method relies on the partitioning of a DNA sample into ~20,000 water–oil emulsion droplets and performing of PCR on each individual droplet containing one or a few copies of the target DNA fragment (Yang et al., 2014). Following PCR amplification, droplets are classified and counted as positive (1) or negative (0), according to the presence or absence, respectively, of the target gene amplification product. Finally, amplified copy numbers of the target gene are calculated as copies/μl based on the Poisson distribution (Hindson et al., 2011). A major advantage of ddPCR over qPCR is that it can be used for absolute target gene quantification without requiring standards, references, or calibration curves (Yang et al., 2014), which is particularly notable when reference material is not available. Moreover, the ddPCR method is a potentially more powerful tool compared to conventional PCR and qPCR due to its greater sensitivity, specificity, accuracy, and reproducibility, and lesser susceptibility to the effects of PCR inhibitors (Huggett et al., 2013; Whale et al., 2013; Yang et al., 2014).
Recently, the ddPCR method has been applied successfully to the quantification of viruses, bacteria, and fungi in medical research, specific pathogen detection in clinical applications, food safety inspection, and gene-editing frequency studies (Cao et al., 2020; Floren et al., 2015; Mock et al., 2016; Taylor et al., 2015; Watanabe et al., 2015). This study was performed to develop a specific and sensitive ddPCR method for the quantification of E. pyrifoliae, and to evaluate its sensitivity using different templates (i.e., genomic DNA, cultured cells, and total DNA from plant tissues). To our knowledge, this is the first report to describe the rapid, sensitive, and absolute quantification of E. pyrifoliae by ddPCR.
Bacterial cells were initially stored in 30% (v/v) glycerol at −80°C and recovered on Luria-Bertani (LB) agar plates at 28°C. After 24 h, a single colony from each strain was selected and inoculated into 10 ml liquid LB with rotation at 180 rpm at 28°C for 12 h. Aliquots of 1 ml bacterial suspensions were centrifuged at 12,000 ×g for 20 min, and the pellets were harvested for DNA extraction. Genomic DNA of all bacteria was extracted using an APrep Total DNA kit (GenomicBase, Seoul, Korea) according to the manufacturer’s instructions. The quality and quantity of isolated genomic DNA were evaluated by spectrophotometry (NanoPhotometer NP80, Implen, Munich, Germany). The concentration of DNA (ng/μl) was converted to copies of DNA (copies/μl) using the formula reported by Pan et al. (2020) and Papić et al. (2017), and DNA was serially diluted to the range of 101–108 copies/ml.
To design a specific primer/probe set, we compared the genomic sequences of three E. pyrifoliae strains (strain: GenBank nos. EpK1/15: CP023567, DSM12163: FN392235, and EpK1/96: FP236842) with those of 56 other species, including Erwinia spp. and different genera using BPGA (Bacterial Pan Genome Analysis Pipeline) v. 1.3 software (Chaudhari et al., 2016). Among several unique sequence regions, the locus EPYR_00057 encoding a hypothetical protein in E. pyrifoliae DSM12163 (CPI84_18825 of strain EpK1/15 and EpC_00550 of strain Ep1/96) was chosen (Supplementary Fig. 1). To evaluate the specificity of this region, the sequence was blasted against the NCBI database. The results confirmed that only E. pyrifoliae contained this region. A specific primer/probe set targeting the locus EPYR_00057 was designed: Pyr-F, 5′-CGGCGCGGGATTTATGTAT-3′; Pyr-R, 5′-CCATGCTGCGTTAGTTGATATTG-3′ and 5′-FAM-AGAAGACTATCAGCGGGAAGCCTACA-BHQ-1-3′. The primers were predicted to produce an amplicon of 106 bp (Supplementary Fig. 1). In silico analysis using the NCBI primer BLAST tool (https://www.ncbi.nlm.nih.gov/tools/primer-blast/) showed that the designed primers were specific for E. pyrifoliae, and no matching sequence was found in any other genus or species. We further tested primer specificity using conventional PCR and qPCR analyses with DNA templates of E. pyrifoliae and six other bacterial strains isolated from apple and pear trees. The results confirmed that the Pyr-F/Pyr-R primer set specifically amplified target gene products from only E. pyrifoliae (Fig. 1).
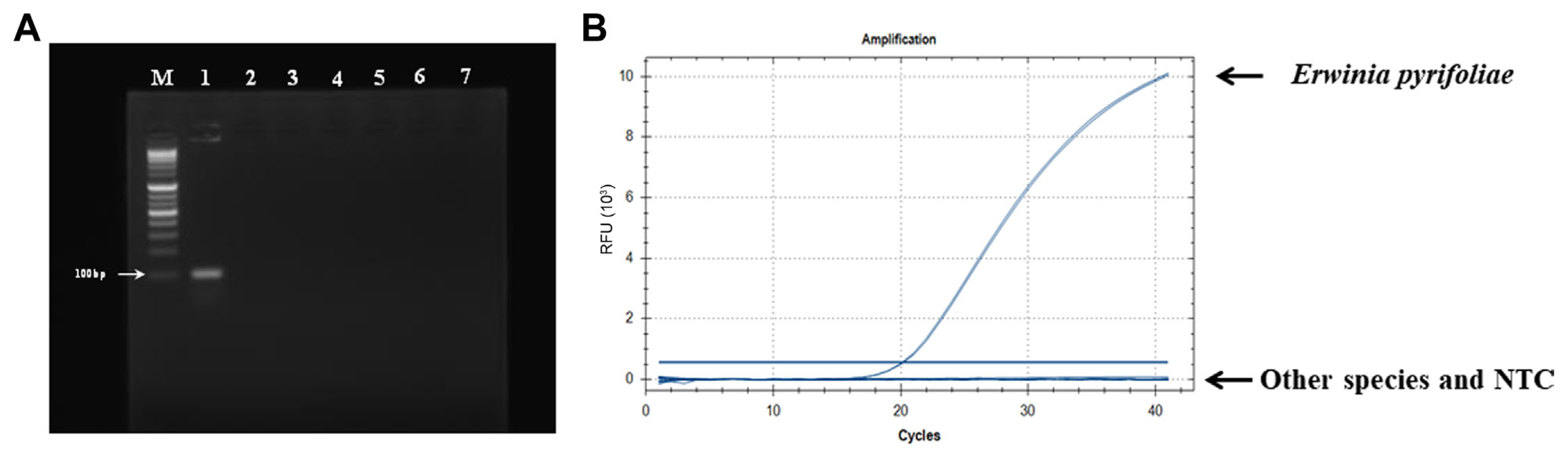
Specific detection of Erwinia pyrifoliae using the species-specific primer pair Pyr-F/Pyr-R. (A) Results of conventional polymerase chain reaction (PCR). Lane M, 1 kb DNA ladder; lane 1, Erwinia pyrifoliae; lane 2, Erwinia amylovora; lane 3, Erwinia billingiae; lane 4, Pantoea agglomerans; lane 5, Pseudomonas viridiflava; lane 6, Pseudomonas graminis; lane 7, Pseudomonas syringae pv. syringae. (B) Specificity of quantitative real-time PCR for detection of E. pyrifoliae. Other species including E. amylovora, E. billingiae, P. agglomerans, P. viridiflava, P. graminis, and P. syringae pv. syringae were not amplified. The negative template control (NTC) used sterile water. RFU, relative fluorescence unit.
ddPCR was performed on a QX200 Droplet Digital PCR system (Bio-Rad, Richmond, CA, USA). PCR was performed in a volume of 20 μl containing 10 μl 2× ddPCR Supermix for Probe (Bio-Rad), 1 μl of each primer (0.3 μM), 0.5 μl (0.25 mM) of the probe, 2 μl genomic DNA or cell culture template, and 5.5 μl ddH2O. The mixtures were transferred into the wells of a DG8 cartridge (Bio-Rad), and 70 μl QX200 droplet generation oil (Bio-Rad) was added via oil holes. After their generation, the droplets were transferred into 96-well PCR plates and sealed using a PX1 plate sealer (Bio-Rad). PCR was performed in a C1000 thermal cycler (Bio-Rad) with the following thermal cycling conditions: 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and annealing at 58.5–65°C for 60 s, 98°C for 10 min, and holding at 4°C, with a ramp rate of 2.0°C/s. After the reaction, the plates were transferred to a QX200 droplet reader (Bio-Rad) and the data were analyzed using QuantaSoft software (Bio-Rad).
Improper PCR conditions, such as an inappropriate annealing temperature, can lead to the emergence of intermediate fluorescent droplets, which would lead to the generation of inaccurate results. Thus, we conducted thermal gradient PCR to determine the optimal annealing temperature using the same amount of genomic DNA and cell culture with the following temperatures: 65°C, 64.5°C, 63.7°C, 62.5°C, 61°C, 59.8°C, 58.5°C, 55.9°C, and 58.5°C. At temperatures from 65°C to 62.5°C, positive (blue) and negative (grey) droplet regions were too close to distinguish, and at temperatures from 61°C to 55.9°C, the degree of rain (i.e., detection of multiple non-specific signals and impaired droplets in the range of positive and negative clusters) in the intermediate region increased (Fig. 2). The results implied that the optimum annealing temperature of the primer/probe designed for the detection of E. pyrifoliae was 62.5°C, which resulted in clear separation of positive from negative droplet clusters with a minimum degree of rain in the genomic DNA (Fig. 2A) and cell cultures (Fig. 2B) of E. pyrifoliae. The sensitivity of ddPCR was evaluated using serially diluted genomic DNA and cell cultures ranging from 101 to 108 copies/ml or cfu/ml. The lower limit of detection of genomic DNA was 3.74 ± 0.04 log copies/ml, and that of cell cultures was 3.82 ± 0.05 log cfu/ml. The upper limit of detection of genomic DNA was 7.11 ± 0.73 log copies/ml and that of cell cultures was 7.21 ± 0.02 log cfu/ml. These data suggested that the detection ranges of E. pyrifoliae in the genomic DNA and cell culture templates were 103–107 copies/ml and cfu/ml, respectively (Fig. 3A).
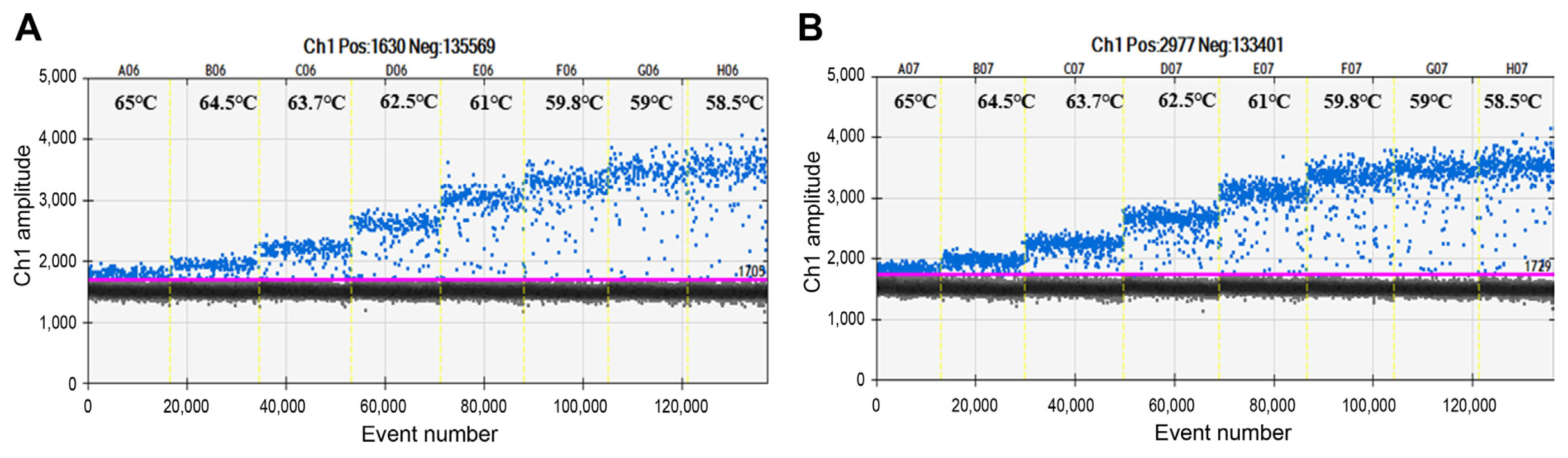
The optimum annealing temperature for the droplet digital polymerase chain reaction (ddPCR) assay. Thermal gradient polymerase chain reaction was run to establish the optimal temperature using genomic DNA (A) and cell culture (B) as the template. The temperature gradient ranged from 58.5°C to 65°C are divided by vertical dotted yellow lines; 62.5°C was further selected by proper separation and reduced rain. The pink line is the threshold, above which are positive droplets (blue) of Erwinia pyrifoliae and below that are negative droplets (gray). The x and y axes represent the number of droplets and fluorescence signal amplitude, respectively.
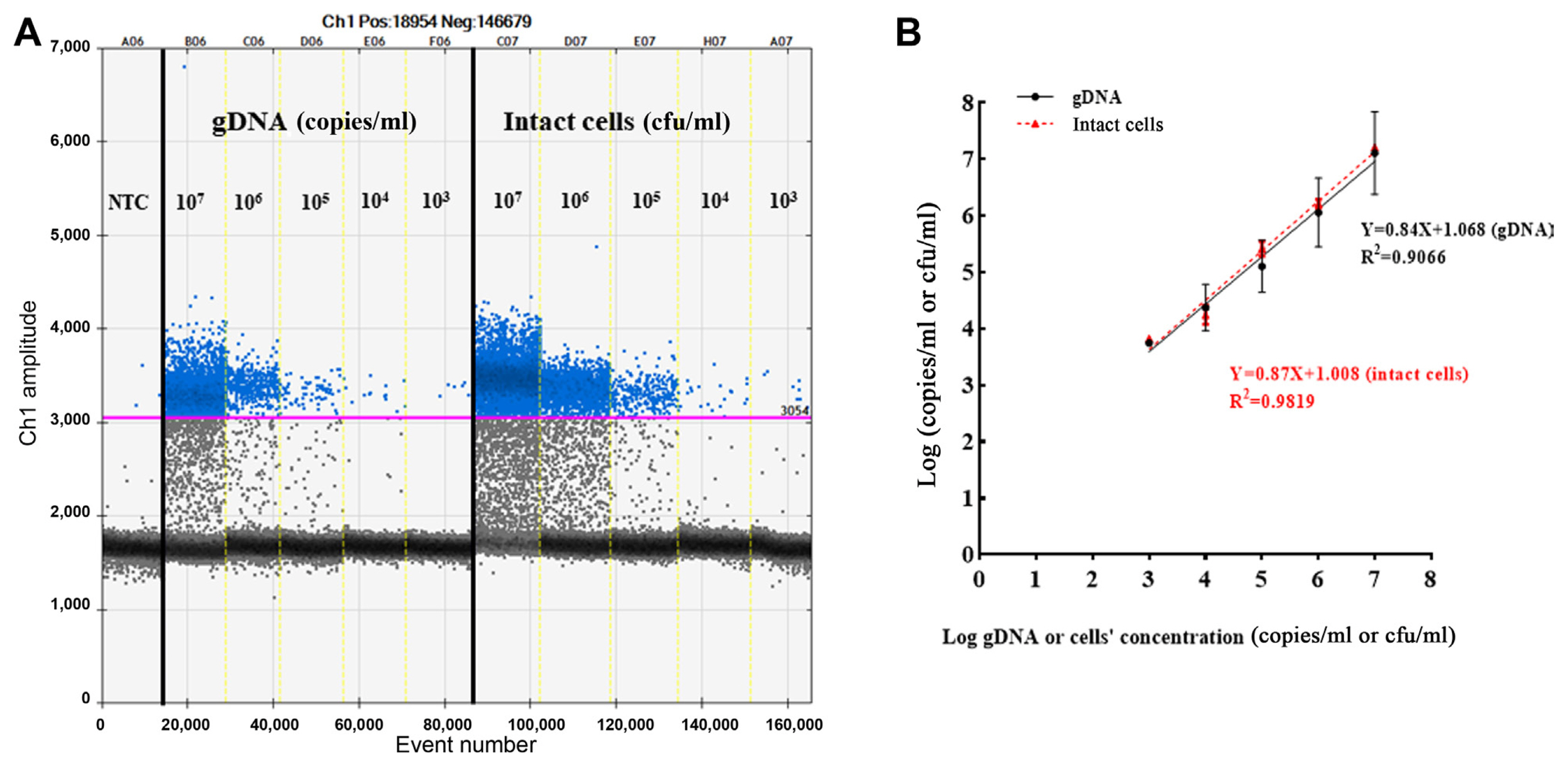
Evaluation of the limit of detection and linear regression of the droplet digital polymerase chain reaction (ddPCR) assay. (A) Limit of detection determined using continuous 10-fold dilutions of genomic DNA and cell culture templates. (B) Linear relationship between the log-transformed values of genomic DNA or bacterial concentration and the corresponding measured values obtained from ddPCR. The linear regression equation and correlation coefficient (R2) are shown on the graph, and the error bars represent the standard deviation from two replicates. NTC, negative template control.
To further investigate the linearity and dynamic range, regression curves of the two templates were constructed. The genomic DNA and cell culture templates showed good correlation (R2 = 0.91 and 0.98, respectively) (Fig. 3B). For repeatability analysis (i.e., intra-assay variation), the E. pyrifoliae suspension was divided into identical quadruplicates, and the reactions were conducted under the same conditions. Finally, the fluorescence signal was evaluated. The results showed 9,757, 9,936, 11,322, and 10,128 positive events, with a coefficient of variation of 5.95%. There was no significant variation among the numbers of positive droplets detected, indicating excellent repeatability (Fig. 4).
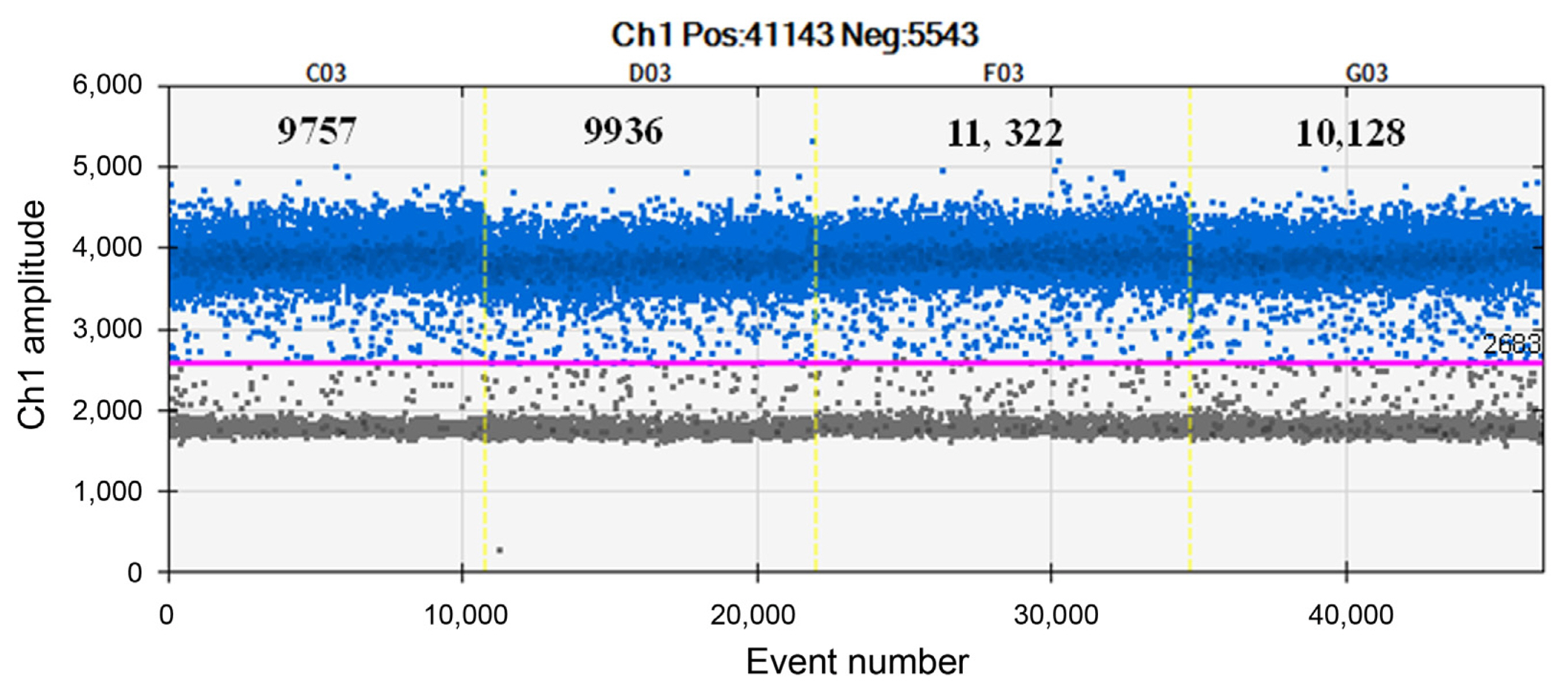
The droplet digital polymerase chain reaction (ddPCR) repeatability assay. Identical suspensions of Erwinia pyrifoliae were divided into quadruplicates for ddPCR detection. The results showed 9,757, 9,936, 11,322, and 10,128 positive events, with a coefficient of variation of 5.95%, indicating excellent repeatability. The pink line is the threshold. Blue and gray dots indicate positive droplets with amplification and negative droplets without amplification, respectively.
To verify the applicability of E. pyrifoliae detection in plant samples, immature pear fruits were inoculated with 10 μl of bacterial suspension (101–104 cfu/ml) using syringes. The negative control was inoculated with sterile water. The inoculated fruits were further incubated in a dark growth chamber (28°C, 90% humidity). Water-soaking and browning necrotic symptoms appeared at 4 days after inoculation on fruitlets inoculated with 103 and 104 cfu/ml of E. pyrifoliae (Fig. 5A). However, minor water-soaking symptoms developed on fruitlets inoculated with 102 cfu/ml of E. pyrifoliae and no disease symptoms developed on fruitlets inoculated with 101 cfu/ml of E. pyrifoliae (Fig. 5A). To quantify the bacteria in plant tissues, 0.2 g tissue slices were cut from symptomatic pear fruits and ground in 1 ml sterile distilled water. Total DNA was extracted from each sample as described above and ddPCR was performed using 2 μl of total DNA as template in each reaction. The results showed that 4.95 × 105 copies/ml of bacterial DNA were detected in asymptomatic tissues inoculated with 101 cfu/ml of E. pyrifoliae suspension, and 3.66 × 106 copies/ml of bacterial DNA were detected in minor water-soaked tissues inoculated with 102 cfu/ml of E. pyrifoliae. The DNA concentration of necrotic tissues inoculated with 103 cfu/ml of bacterial suspension was 5.37 × 107 copies/ml of bacterial DNA. E. pyrifoliae was positively detected in severely diseased tissues inoculated with 104 cfu/ml of bacterial suspension, but all the fluorescence signal amplitudes were positive, implying that the levels exceeded the upper detection limit of ddPCR (Fig. 5B). No E. pyrifoliae was detected in the negative control, which confirmed that ddPCR is suitable for black shoot blight disease detection even in asymptomatic tissues.
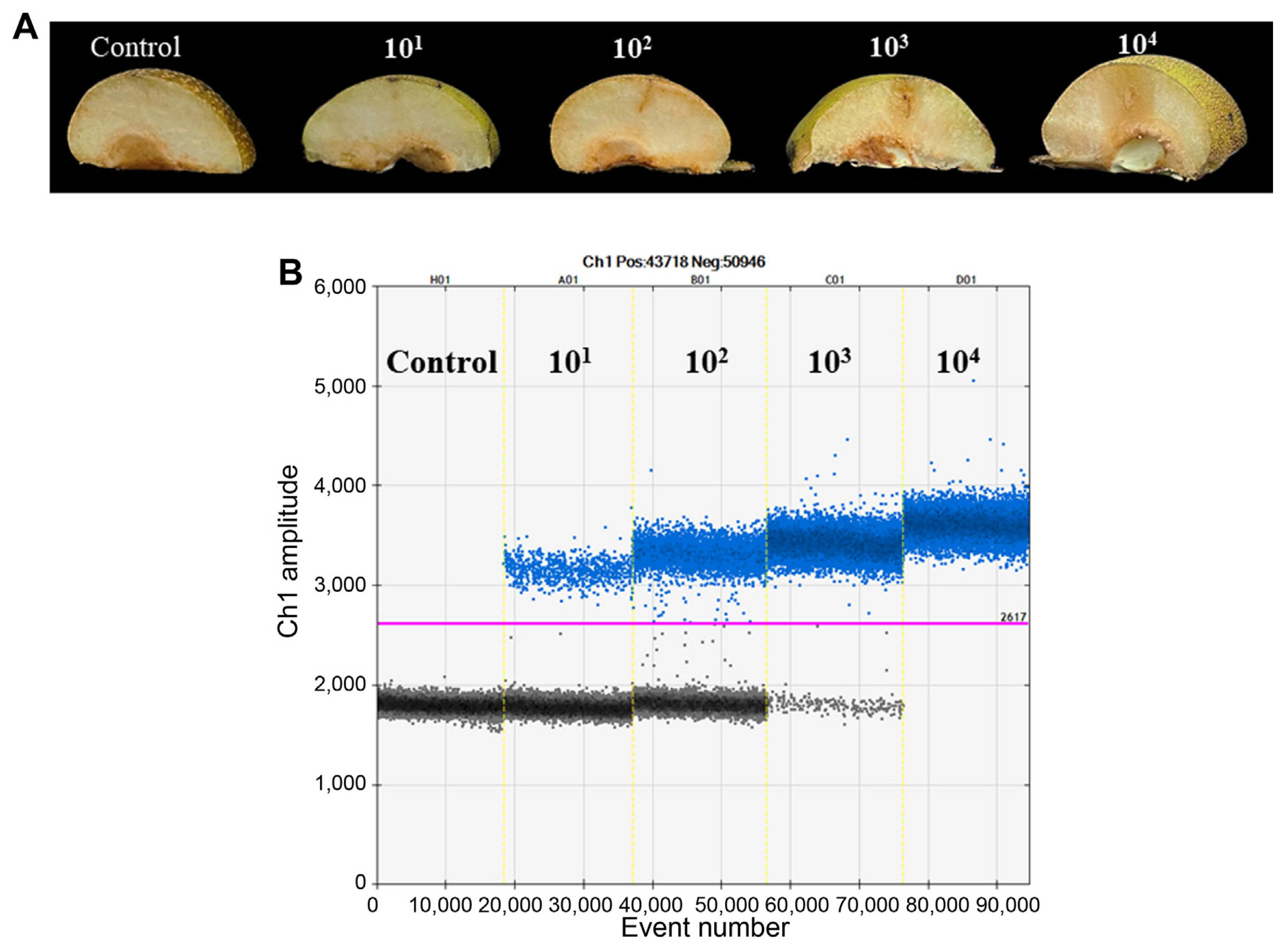
Detection of Erwinia pyrifoliae from artificially inoculated immature pear fruits. (A) Symptoms of immature pears inoculated by continuous 10-fold dilution of bacterial suspension (101–104 cfu/ml). The negative control was inoculated with sterile water. (B) Droplet digital polymerase chain reaction assay for species-specific detection of E. pyrifoliae in total DNA extracted from artificially inoculated immature pear fruits. The pink line is the threshold. Blue and gray dots indicate positive droplets with amplification and negative droplets without amplification, respectively.
As an emerging technology, ddPCR has significant potential to improve the current practice of using other PCR-based methods (e.g., conventional PCR and qPCR) for molecular-based detection and quantification. In the field of plant pathology, ddPCR technology has been developed and applied widely for the sensitive detection and absolute quantification of various plant pathogens since the first reporting of its application for plant pathogenic bacteria (Dreo et al., 2014; Wang et al., 2022). In the present study, we established a ddPCR method for the detection and quantification of E. pyrifoliae using genomic DNA and cell culture templates. To improve the method’s specificity, we developed a highly specific primer/probe set to target a unique gene in the E. pyrifoliae DSM12163 genome (EPYR_00057, hypothetical protein). The ddPCR method showed high sensitivity in vitro, with a lower detection limit of 103 (5.5 × 103 copies/ml and 6.7 × 103 cfu/ml) and an upper limit of 107 (4.24 × 107 copies/ml and 1.6 × 107 cfu/ml) in genomic DNA and cell culture PCR templates, respectively. We also tested ddPCR for the detection of E. pyrifoliae from plant tissue samples. The results showed that 4.95 × 105 copies/ml of bacterial DNA were detected despite the lack of symptomatic tissues. Although the number of bacteria exceeded the upper limit of quantification in severe lesions, E. pyrifoliae was still identified based on the specific primer and probe. These results indicate that, ddPCR is suitable for the accurate diagnosis of E. pyrifoliae.
Several qPCR-based methods and specific primers have been developed for the quantification of E. pyrifoliae (Jin et al., 2022; Lehman et al., 2008; Wensing et al., 2012). The sensitivity of our ddPCR method was similar to or better than that of these previously reported techniques. Several recent studies have yielded similar results, with similar limits of detection, for qPCR and ddPCR methods (Dreo et al., 2014; Dupas et al., 2019). ddPCR has the major advantage of the ability to absolutely quantify target copies calculated by Poisson’s distribution without reference material (Huggett et al., 2013; Whale et al., 2013; Yang et al., 2014). In contrast, qPCR methods involve the estimation of target copy numbers based on Cq values and a standard curve, which may result in random errors or quantification bias (Gutiérrez-Aguirre et al., 2015; Sahu et al., 2021). In addition, ddPCR appears to be less susceptible to PCR inhibitors derived from crude samples of plant materials or soil (Gutiérrez-Aguirre et al., 2015; Wang et al., 2022). In conclusion, we established a rapid and absolute quantitative detection method for E. pyrifoliae using bacterial genomic DNA and cell culture. This ddPCR method is a promising tool for application in the field of plant pathology or epidemiological surveys for plant quarantine agencies and plant clinics.
Acknowledgments
We thank Dr. Sunghoon Jung for the use of laboratory equipment, and Dr. Mi-Hyun Lee for providing E. amylovora genomic DNA. This project was supported by the Animal and Plant Quarantine Agency (PQ20204B026), Ministry of Agriculture, Food and Rural Affairs, Republic of Korea.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).