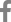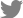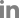Ixora coccinea, commonly known as jungle geranium, is a flowering shrub native to the countries of Eastern South Asia, such as India, Bangladesh, and Sri Lanka, and it is now cultivated in both tropical and subtropical countries. In addition, it is used as a medicinal plant for skin disease, dysentery, ulcers, and gonorrhea (Shreelakshmi et al., 2021). With the increasing demand, the import and cultivation of I. coccinea has been growing in South Korea, and this is mostly propagated by cuttings (Lin et al., 2020). Although several studies reported natural infections of fungal pathogens, including Mycoleptodiscus indicus and Colletotrichum aeschynomenes, in I. coccinea (Banerjee et al., 2018; Li et al., 2021), no viral pathogens have been reported to date.
Recently, Illumina high-throughput sequencing-based diagnostic platforms have been widely utilized to identify various unknown viral pathogens in crops (Ibaba and Gubba, 2020; Rivarez et al., 2021). In addition to its advantages, this technology also has some limitations in viral diagnosis, including costly equipment, long sequencing times, low portability, and complex data analysis (Fitzpatrick et al., 2021). Nanopore sequencing introduced by Oxford Nanopore Technology (ONT), known as third-generation sequencing, has several advantages compared to Illumina-based sequencing, including reduced cost, high portability, single-molecule (DNA or RNA) sequencing, long sequencing reads, faster sequencing, and easy data analysis that can be performed on a personal computer, which facilitate its use in plant viral diagnostics (Lee et al., 2022; Sun et al., 2022). The biggest drawbacks of nanopre sequencing have been the lower throughput of sequence data and an approximate 10% high error rate. However, recent research has achieved validation of the accuracy of nanopore sequencing long reads through improvements in technology (Lu et al., 2016).
In September 2022, two out of the 20 I. coccinea plants showing foliar disease symptoms with mosaic and mild mottle were collected from the streets of Jeju Island, South Korea, where they had been displayed (Fig. 1). Total RNA was extracted from symptomatic leaf samples (~200 mg) using a Clear-S Total RNA extraction kit (Invirustech Co., Gwangju, Korea) according to the manufacturer’s instructions. DNA removal and RNA purification for library construction were performed as described by Lee et al. (2022). The purified RNA was used for cDNA synthesis using a Maxima H Minus Double-Stranded cDNA Synthesis Kit (Thermo Fisher Scientific, Waltham, MA, USA). For library construction, dA tailing and adapter ligation were performed using a NEBNext Ultra II End Repair/dA-Tailing Module (New England Biolabs, Ipswich, MA, USA) and a Blunt/TA Ligase Master Mix (New England Biolabs) with an Adapter Mix (SQK-DCS109, Oxford Nanopore Technologies, Oxford, UK), respectively, generating the nanopore library with a final concentration of 3.3 ng/μl. The procedures of sample loading into Flongle flow cell (R9.4.1), sequencing of the nanopore library, and subsequent analyses of the reads were performed as described by Lee et al. (2022). For metatranscriptomics analysis, the FASTQ files were converted to FASTA files, and the adapter sequences were removed using the tool Geneious Prime (version 21.1.1, Biomatters, Auckland, New Zealand). The FASTQ data files were deposited in the National Center for Biotechnology Information (NCBI) Sequence Read Archive repository under SRR23561814. To identify genome sequences of plant viruses, ONT EPI2ME WIMP workflow (version 3.4.2) and BLASTn search on the NCBI Viral Genome data (version 21.11.4, Viral RefSeq Database, VirDB) were utilized as described by Lee et al. (2022).
The nanopore sequencing generated a total of 434,106 raw reads with an average length of 317.1 nucleotides. After adapter removal and quality filtering steps (>Q7), a total of 349,069 reads with an average length of 44.6 nucleotides were obtained (Table 1). The identification of plant virus-associated sequences was performed using the EPI2ME WIMP workflow. A total of 796 (0.22%) virus-like reads were found and were BLASTed on the NCBI VirDB. All virus-like reads were mapped to jasmine virus H (JaVH) (Table 2).
De novo assembly of MinION reads was performed with genome sequences of JaVH isolate (JaVH-CNU) (Fig. 2A). A total of 796 reads (0.22% of the total reads) were mapped with 95.5% nucleotide coverage and 95.1% nucleotide identity. To obtain the complete genome sequence of JaVH, the consensus sequences were finally mapped against a reference viral genome (NC055545; at 60× coverage depth and at least 20% of support fraction for base-call ambiguity). In addition, Sanger sequencing after reverse transcription polymerase chain reaction (RT-PCR) was performed for filling in any sequence gaps using virus-specific primers (Fwd: 5′-TCC AAG GCG AAT GCT CTC TCT GT-3′/Rev: 5′-GGA TTG TAG TGG AGC GGT GAA AAA CCC-3′), which amplify the fragments between the aligned reads, designed based on the consensus sequence. The RT-PCR reaction mixture (final volume 20 μl) contained 10 μl SuPrimerScript RT Premix (Genet Bio, Daejeon, Korea), 2 μl RNA template, 2 μl of each of the 10 μM forward and reverse primers, and diethylpyrocarbonate-treated water. Amplification reaction was conducted under the following conditions: 50°C for 30 min; 95°C for 5 min; 35 cycles of 95°C for 30 s, 62°C for 30 s, and 72°C for 30 s; and 72°C for 5 min. The 5′ and 3′ end sequences of JaVH were determined by rapid amplification of cDNA end (RACE) technique using a SMARTer RACE 5′/3′ Kit (Clontech, Mountain View, CA, USA). The complete genome consensus sequence of the JaVH infecting I. coccinea was deposited in GenBank (LC757512).
The genome structure of JaVH-CNU was predicted using the ORFfinder software (http://www.ncbi.nlm.nih.gov/orffinder). The RNA genome of JaVH-CNU consisted of 3,867 nucleotides (nt), with a total of four open reading frames (ORFs) (Fig. 2B). The 5′ and 3′ untranslated regions were found to be 19 nt and 226 nt in length, respectively. The first ORF (ORF1) comprised 729 nt and has potential to read-through a downstream ORF, resulting in production of a 2,301 nt sequence encoding the RNA-dependent RNA polymerase (RdRP). The ORF2 (189 nt) and ORF3 (252 nt) encoded movement protein. The ORF4 (1020 nt) encoded a coat protein (CP) (Fig. 2B).
To confirm the presence of JaVH in I. coccinea, RT-PCR was performed using JaVH-specific diagnostic primer sets (Dey et al., 2018), and the RT-PCR product (657 bp) was sequenced by Sanger sequencing (Fig. 2C). The obtained sequences confirmed the presence of JaVH (data not shown).
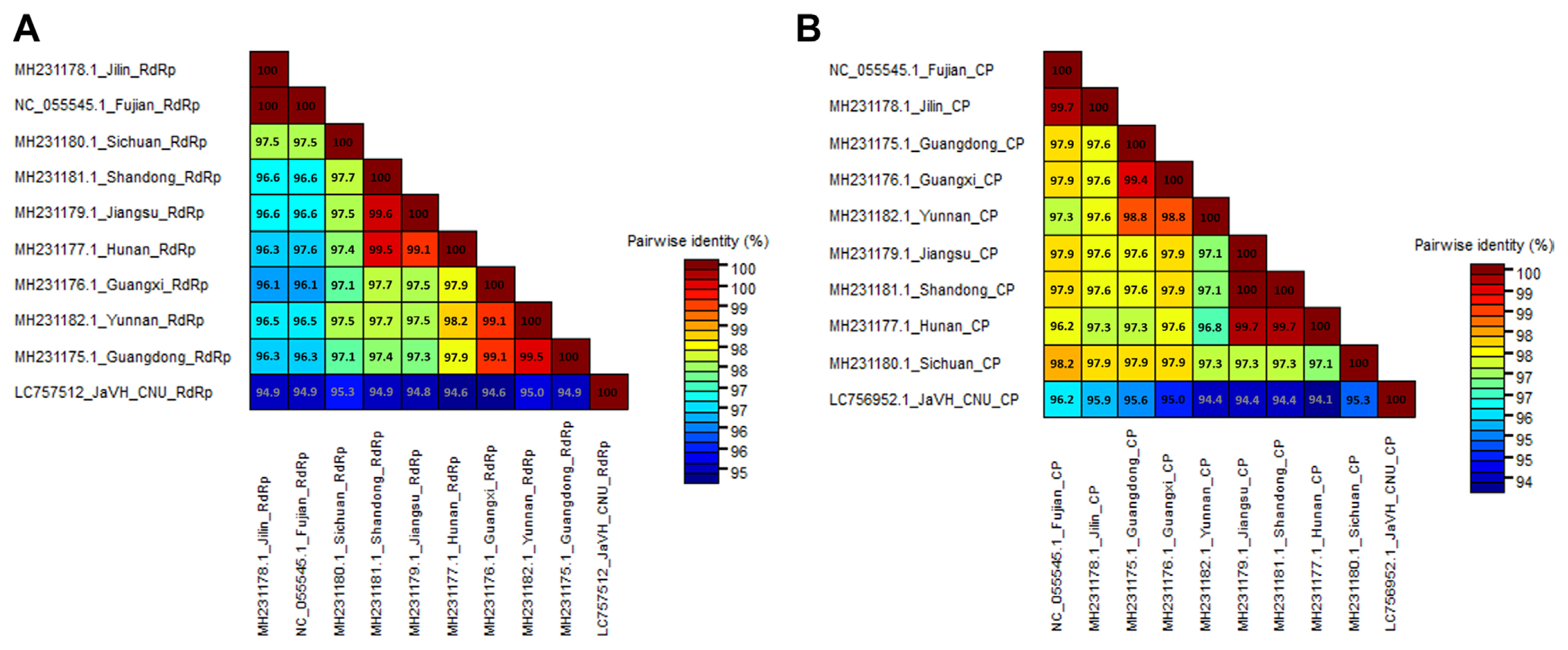
The complete genome of JaVH-CNU exhibited sequence identities ranging from 88.4% (JaVH-Hunan, MH231179) to 90.3% (JaVH-Fujian, NC055545) with other JaVH isolates. In addition, to compare the amino acid sequences, the complete amino acid sequences of RdRP and CP of JaVH-CNU were aligned with those of other JaVH isolates available on GenBank using BioEdit software version 7.2.5 (Ibis Biosciences, Carlsbad, CA, USA). Pairwise distances were calculated using the PASC algorithm (NCBI, Bethesda, MD, USA) available on the GenBank database. JaVH-CNU shared amino acid sequence identities of 94.9-95.3% in RdRP (Fig. 3A) and 94.4-96.2% in CP (Fig. 3B).
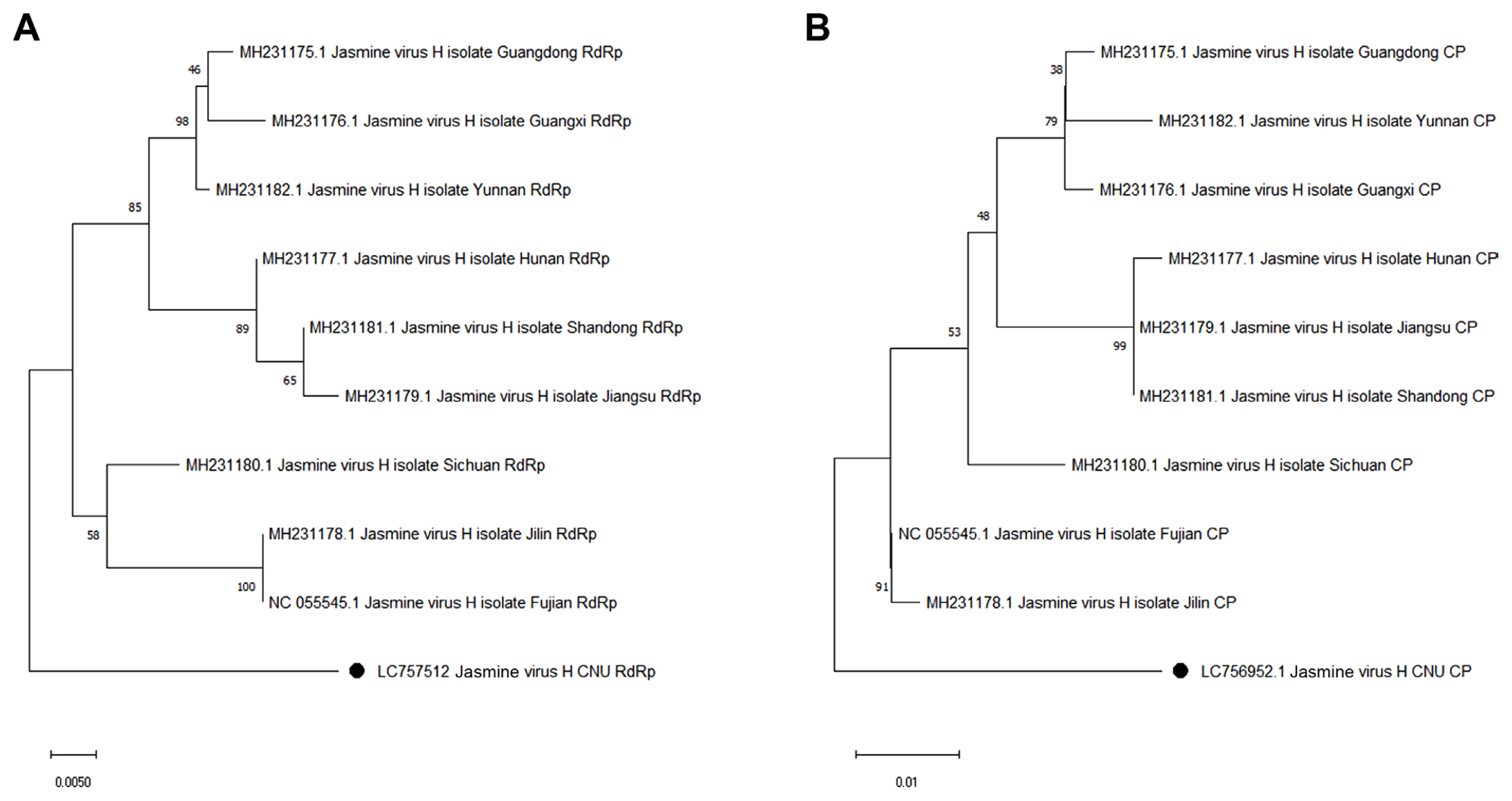
To assess the phylogenetic relationship among JaVH-CNU and other JaVH isolates, the complete amino acid sequences of the RdRP and CP of JaVH-CNU, along with those of other JaVH isolates, were used in phylogenetic analysis. The phylogenetic tree was constructed using the MEGA 10.0 tool (Kumar et al., 2018). The alignments were used to infer neighbor-joining trees in MEGA 10.0 with Tamura-Nei model and 1,000 bootstrap replicates as described by Lee et al. (2022). Phylogenetic analysis of RdRP and CP revealed that the JaVH-CNU differed from the existing isolates in GenBank. The RdRP and CP of JaVH-CNU were clustered separately with different JaVH isolates, probably having different biological characteristics (Fig. 4).
The spread of plan viruses through global trade is a significant concern. The international movement of infected plant materials or their products can introduce viruses to new geographic locations and host plant species, potentially leading to the emergence of new disease. Moreover, the movement of insect vectors, such as aphids or whiteflies, can also facilitate the spread of plant viruses through global (Amari et al., 2021). To successfully respond to an unexpected outbreak of plant viruses, it has become increasingly important to establish rapid and reliable diagnostic methods for effective disease management.
With the advantages of nanopore sequencing, this study sequenced and identified the full genome of a JaVH isolate from I. coccinea, and it took only 48 h from RNA extraction to virus identification in the laboratory, providing rapid diagnosis for disease survey and management. The JaVH, which has been recently characterized from Jasminum sambac (Zhuo et al., 2017) and J. multiflorum (Dey et al., 2018), belongs to a member of the genus Pelarspovirus in the family Tombusviridae. Despite the JaVH isolates reported in other countries and hosts, the genome of JaVH seems to be highly variable based on phylogenetic analyses of complete amino acid sequences of RdRP and CP (Dey et al., 2018). It is worth noting that this is first report of a natural infection of JaVH in I. coccinea worldwide. Thus, it is urgently required to survey the incidence of JaVH in I. coccinea in Korea, and more importantly, to evaluate the disease risk in different types of crops and ecologies.

 PDF Links
PDF Links PubReader
PubReader ePub Link
ePub Link Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print