Viral Metatranscriptomic Analysis to Reveal the Diversity of Viruses Infecting Satsuma Mandarin (Citrus unshiu) in Korea
Article information

Abstract
Citrus cultivation plays a pivotal role, making a significant contribution to global fruit production and dietary consumption. Accurate identification of viral pathogens is imperative for the effective management of plant viral disease in citrus crops. High-throughput sequencing serves as an alternative approach, enabling comprehensive pathogen identification on a large scale without requiring pre-existing information. In this study, we employed HTS to investigate viral pathogens infecting citrus in three different regions of South Korea: Jeju-do (Jeju), Wando-gun (Wando), and Dangjin-si (Dangjin). The results unveiled diverse viruses and viroids that exhibited regional variations. Notably, alongside the identification of well-known citrus viruses such as satsuma dwarf virus, citrus tatter leaf virus, and citrus leaf blotch virus (CLBV), this study also uncovered several viruses and viroids previously unreported in Korean citrus. Phylogenetic analysis revealed that majority of identified viruses exhibited the closest affilations with isolates from China or Japan. However, CLBV and citrus viroid-I-LSS displayed diverse phylogenetic positions, reflecting their regional origins. This study advances our understanding of citrus virome diversity and regional dynamics through HTS, emphasizing its potential in unraveling intricate viral pathogens in agriculture. Consequently, it significantly contributes to disease management strategies, ensuring the resilience of the citrus industry.
Citrus is a genus of flowering plants in the Rutaceae family, which is commonly known as the citrus family. These plants are renowned for their fruit-bearing trees and shrubs, which produce a wide variety of citrus fruits. Citrus fruits are known for their bright, zesty flavors, and they are widely consumed around the world due to their refreshing taste and nutritional value (Acosta et al., 2023). Two hundred million tons of citrus fruits are produced annually from cultivated areas worldwide (Food and Agriculture Organization of the United Nations, 2020). Citrus varieties or cultivars refer to the many distinct types of citrus fruits that have been developed through selective breeding or natural mutations over the years. These cultivars such as Navel oranges (Citrus × sinensis ‘Washington’ or ‘Riverside’), Valencia oranges (Citrus × sinensis ‘Valencia’), Meyer lemons (Citrus × meyeri), and Satsuma mandarins (Citrus unshiu) can vary in terms of flavor, appearance, and other characteristics (Zibaee et al., 2020). Notably, citrus production in Korea, particularly on Jeju-do, is a significant agricultural industry. The country’s climate and geographical conditions, including its volcanic soil and mild winters, make it suitable for growing various citrus fruits. The reported citrus production for Korea, however, consists only of tangerines and mandarins. In 2022, citrus production totaled 595,000 metric tons (MT) in 19,930 hectares, which amounted to approximately 1 billion USD (http://www.krei.re.kr).
Despite the tremendous extent and value of citrus production, the quality and yield of citrus can be affected by several viral diseases that pose significant challenges to growers. There are several major citrus viruses that can impact citrus trees and thus the citrus industry. These viruses, including citrus tristeza virus (CTV), citrus exocortis viroid (CEVd), citrus tatter leaf virus, citrus psorosis virus, and citrus ringspot virus, can cause various symptoms and pose significant challenges to growers (Bester et al., 2021). Additionally, the transmission of citrus viruses primarily involves vectors such as aphids, nematodes, grafting, and propagation, indicating that different citrus viruses have different vectors (Yokomi, 2019). Recently, several viruses, including citrus leaf blotch virus (CLBV), CLBV2, and citrus vein enation virus, have been reported in Satsuma mandarins in Korea (Kwak and Kil, 2022; Park et al., 2019; Yang et al., 2019). However, no research has been conducted on a nationwide scale to investigate the virus communities distributed throughout the country. Currently, citrus viral disease diagnosis is frequently performed by nucleic acid-based molecular detection methods such as reverse transcription-polymerase chain reaction (RT-PCR) and TaqMan probe-based reverse transcription quantitative PCR (Hyun et al., 2017; Kokane et al., 2021). However, these PCR-based diagnostic techniques are not always adequate for the simultaneous detection and identification of multiple citrus viruses and unknown viruses. Due to the vast diversity of the virosphere, these approaches often struggle to identify new virus species or strains in symptomatic citrus samples (Villamor et al., 2019).
High-throughput sequencing (HTS), also known as next-generation sequencing, is a powerful technology that has revolutionized genomics and molecular biology and is used for rapidly and efficiently determining the nucleotide sequences of DNA or RNA molecules. Its principles involve preparing nucleic acid libraries, sequencing the genetic material, and then analyzing and interpreting the resulting data to extract valuable biological information (Villamor et al., 2019). It plays a pivotal role in the monitoring and management of viral diseases in agriculture, thus safeguarding crop yields and food security.
In this study, we employed HTS to identify and characterize viruses infecting Satsuma mandarins (C. unshiu), which is one of the most popular citrus cultivars in Korea, as well as examine the virosphere in major citrus cultivation areas of Korea. This HTS-based viral metatranscriptomic analysis offers a valuable approach for the prevention and control of viral diseases in citrus crops.
Materials and Methods
Sample collection and RNA extraction
In June 2022, citrus (C. unshiu) plants exhibiting symptoms of virus-like diseases were collected from three regions in South Korea: Jeju, Wando, and Dangjin. The collected samples included 211 from Jeju, 126 from Wando, and 32 from Dangjin, which were then pooled for analysis. The total RNA from the pooled citrus samples for each region was extracted using the Clear-S Total RNA extraction kit (InVirusTech Co., Gwangju, Korea), which involved an on-column DNase treatment, following the manufacturer’s instructions. Subsequently, the quality and quantity of the extracted total RNA were assessed using a spectrophotometer (BioDrop, Biochrom Ltd., Cambridge, UK) and the Qubit 4 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). The extracted RNA was then stored at −80°C until further use.
Library preparation for RNA-Seq and virus identification
The DNA-free total RNA underwent ribosome depletion with the Ribo-zero rRNA removal kit for plants (Illumina, San Diego, CA, USA). Subsequently, the cDNA libraries were prepared from ribosome-depleted RNA using the TruSeq Stranded mRNA LT Sample Prep kit (Illumina). The size and quality of the cDNA libraries were then assessed using the Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA). Paired-end Illumina sequencing was conducted on the cDNA libraries using the Illumina NovaSeq 6000 (Illumina), generating 151 base pair strand-specific reads. The bcl2fastq program was then employed to demultiplex the multiplexed sample data. Prior to mapping, the FastQC program was utilized for throughput and quality checks. Data with low quality, defined as having Phred scores lower than or equal to Q20 and containing adapter sequences, were removed. The trimmed paired-end RNA-Seq reads were then mapped against the National Center for Biotechnology Information (NCBI) plant virus database using the Kallisto program (https://pachterlab.github.io/kallisto/). For aligning the reads to the identified plant virus genome, the ‘Map to Reference’ function in Geneious Prime 2022.2.2 was employed with default settings (Medium Sensitivity/Fast), resulting in the generation of consensus sequences.
Phylogenetic analysis
The phylogenetic analysis utilized identified viral species, relying on coat protein (CP) sequences or complete sequences (in the case of viroids) obtained from confirmed plant viruses identified through the Illumina platform. Sequence alignment was conducted with ClustalW multiple alignment using the BioEdit Sequence Alignment Editor (version 7.0.5.3). Phylogenetic analysis was executed through the maximum-likelihood method in MEGA X (version 10.2.4), employing the Tamura-Nei model. Bootstrap values were calculated based on 1,000 iterations.
Analysis of single nucleotide polymorphisms
In this study, single nucleotide polymorphism (SNP) analysis was performed on viruses and viroids with complete or nearly complete sequences. SNPs were identified by mapping the data to the reference genome obtained through genome assembly, utilizing the ‘Find variations/SNPs’ feature in the Geneious Prime software.
Validation of the identified viruses using RT-PCR
The presence of viruses identified by the Illumina platform was validated through RT-PCR. Specific primer sets and the Superscript RT-PCR premix (Genet Bio, Daejeon, Korea) were employed for RT-PCR, following the manufacturer’s instructions.
Data availability
The FASTQ files generated in this study have been deposited in the NCBI Sequence Read Archive (SRA) repository under the accession numbers SRR23239774 (Ci-JJ), SRR23239773 (Ci-WD), and SRR23239772 (Ci-DJ). These files are also accessible in the NCBI BioProject database, assigned the accession number PRJNA928104. Furthermore, the sequences of the identified plant viruses have been submitted to the NCBI GenBank with individual accession numbers.
Results
Library construction and virus identification
Total RNA was extracted from citrus leaves with virus-like disease symptoms in three regions (Jeju, Wando, and Dangjin), and libraries were prepared (Fig. 1A and B). Metatranscriptome data were then generated through paired-end sequencing (Table 1). The sizes of the raw RNA-Seq data obtained through sequencing were 6.1 Gb, 6.7 Gb, and 7.0 Gb for the Jeju, Wando, and Dangjin samples, respectively, and have been deposited in the SRA database. The number of reads obtained was 40,703,166 from the Jeju samples, 44,958,884 from the Wando samples, and 46,927,060 from the Dangjin samples. Contigs were generated through data trimming and de novo assembly, resulting in 98,361 contigs from the Jeju samples, 96,586 from the Wando samples, and 93,167 from the Dangjin samples.
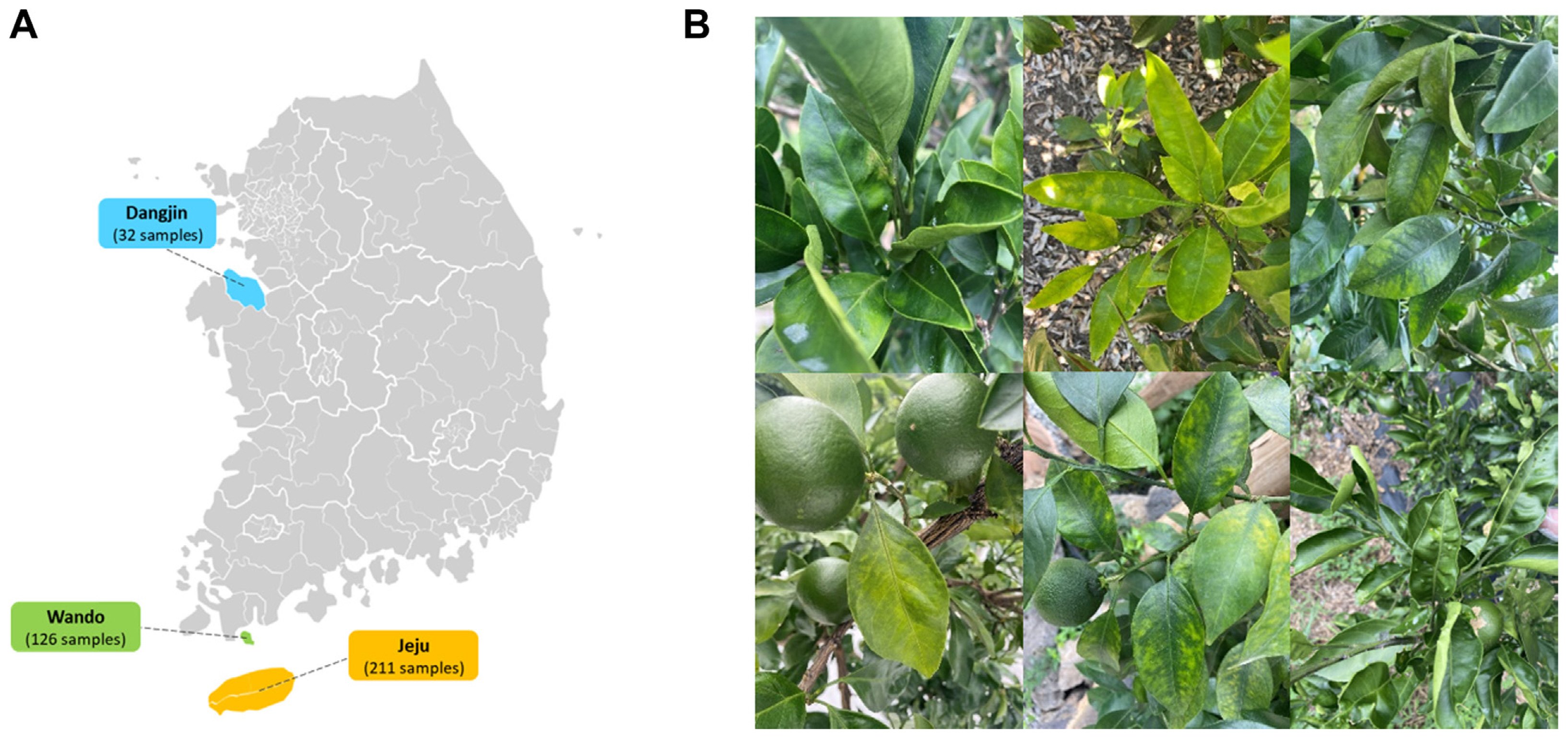
(A) Citrus collection location at region in Korea. (B) Virus-like disease symptoms on Citrus unshiu leaves.
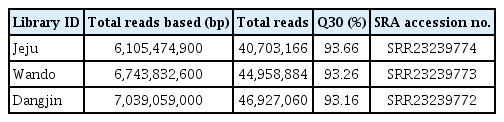
Summary of sequencing data
Identification of viruses and viroids from citrus transcriptomes
To identify the viral reads and contigs, a Nucleotide Basic Local Alignment Search Tool (BLASTN) search against a virus reference genome database from NCBI was conducted using the assembled reads and contigs from each library. Through this BLASTN search, five viruses and five viroids were identified in Jeju, two viruses and six viroids in Wando, and one virus and four viroids in Dangjin. The number of viral reads was 48,570 from the Jeju library, 2,837 from the Wando library, and 2,443 from the Dangjin library, respectively (Fig. 2A). In the Jeju library, the predominant proportion of reads was attributed to CLBV, accounting for 34,497 (71.03%). In the Wando library, citrus exocortis Yucatan viroid (CEYVd) had the highest number of viral reads comprising 962 (33.91%), while in the Dangjin library, citrus viroid-I-LSS (CVd-I-LSS) had the highest proportion with 1,246 reads (51.00%). The identified viruses and viroids were visualized in a heatmap, including their coverage and identities (Fig. 2B). The consensus sequences for each virus and viroid have been deposited in the NCBI database. However, satsuma dwarf virus (SDV) RNA1 and CTV could not be deposited due to low coverage.

(A) The number of reads associated with the identified viruses and viroids in each citrus transcriptome. (B) Heatmap comparing total reads, read number, coverage (%), and identity (%) data. - means no data. CBLVd, citrus bent leaf viroid; CEYVd, citrus exocortis Yucatan viroid; CiMV, citrus bent leaf viroid; CLBV, citrus leaf blotch virus; Cvd-I-LSS, citrus viroid-I-LSS; CVd-III, citrus viroid III; CVd-VI, citrus viroid VI; HSVd, hop stunt viroid; SDV, satsuma dwarf virus.
The amount of virus RNA is determined by normalizing the number of aligned reads to the length of the corresponding gene. To assess this, the Fragments Per Kilobase of transcript per Million mapped reads (FPKM) method was employed (Fig. 3) (Zhao et al., 2021). The FPKM was calculated by dividing the number of aligned reads by the length of the gene, then dividing by the total number of reads and multiplying the result by 109. The virus quantities calculated using the FPKM revealed that in Jeju, CLBV accounted for the highest proportion at 34.4%. Additionally, in Wando, CEYVd was dominant at 43.71%, and in Dangjin, CVd-I-LSS showed the highest abundance at 52.69%.
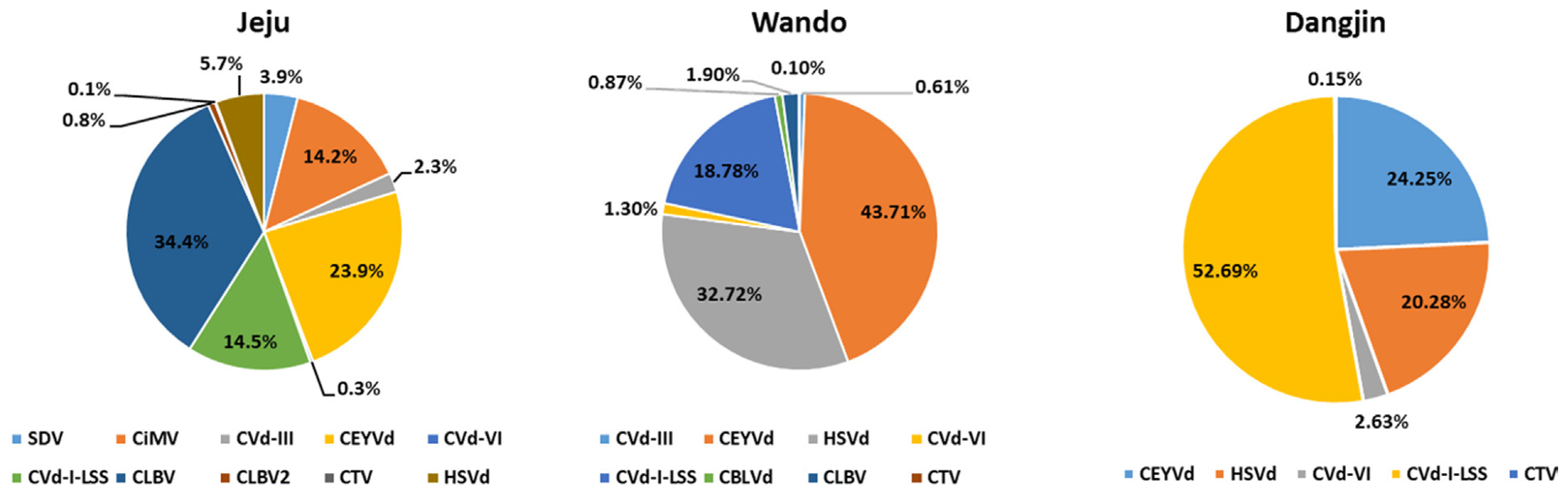
The portion of identified viruses and viroids in the citrus transcriptomes based on Fragments Per Kilobase of transcript per Million mapped reads (FPKM) values. CBLVd, citrus bent leaf viroid; CEYVd, citrus exocortis Yucatan viroid; CiMV, citrus bent leaf viroid; CLBV, citrus leaf blotch virus; CTV, citrus tristeza virus; Cvd-I-LSS, citrus viroid-I-LSS; CVd-III, citrus viroid III; CVd-VI, citrus viroid VI; HSVd, hop stunt viroid; SDV, satsuma dwarf virus.
Genome assembly
The genome assembly was performed for the identified viruses and viroids based on their reference genomes (Fig. 4). In Jeju, 10 identified viruses and viroids exhibited coverage ranging from 39.9% (CTV) to 100%, with identities ranging from 80.8% (CEYVd) to 99.2% (CLBV). In Wando, nine viruses and viroids showed coverage from 21.8% (CTV) to 100% and identities from 88.6% (CEYVd) to 98.9% (CLBV). Finally, in Dangjin, coverage ranged from 50.6% (CTV) to 100%, and the identities varied from 86.5% (CEYVd) to 99.0% (citrus viroid VI [CVd-VI]). Gap-filling was conducted to obtain the complete consensus sequences for citrus mosaic sadwavirus (CiMV), CLBV2, and CVd-VI in the Jeju transcriptome, and for CLBV in the Wando transcriptome (data not shown).

Genome assembly of identified viruses in citrus transcriptomes. (A) A total of 10 viruses and viroids from the Jeju library. (B) A total of eight viruses and viroids from the Wando library. (C) A total of five viruses and viroids from the Dangjin library.
The phylogenetic analysis of the identified viruses and viroids
Phylogenetic analysis was performed to investigate the evolutionary relationships of viruses identified in three regions (Jeju, Wando, and Dangjin) through RNA-Seq (Fig. 5). The CP sequences from the consensus sequences and virus isolates registered in the NCBI database were used for the analysis. The results of the phylogenetic analysis for each identified virus and viroid revealed that the majority exhibited the closest relationships with isolates from China or Japan. Initially, SDV, CLBV2, and CEYVd exhibited close relationships to isolates from China (MT344108 for SDV; MH144344 for CLBV2; and MZ463745 for CEYVd). Furthermore, CiMV, citrus bent leaf viroid (CBLVd), and hop stunt viroid (HSVd) demonstrated high phylogenetic relationships with isolates from Japan (AB465582 for CiMV; AB054636 for CBLVd; and AB054601 for HSVd). Interestingly, CLBV and CVd-I-LSS exhibited different phylogenetic positions depending on the region of the library. CLBV identified in Jeju exhibited a close relationship with isolates from New Zealand (JQ013967), while CLBV from Wando displayed a relationship with isolates from China (MT863785). Regarding CVd-I-LSS, isolates from Jeju and Dangjin exhibited relationships with isolates from China (KF726093), while isolates from Wando were associated with isolates from Japan (AB054638). In contrast to the patterns observed for most viruses and viroids related to isolates from China or Japan, CVd-III showed a unique pattern, with Jeju isolates clustering with isolates from Pakistan (FJ773275), and Wando isolates clustering with isolates from the United States (JX259428).
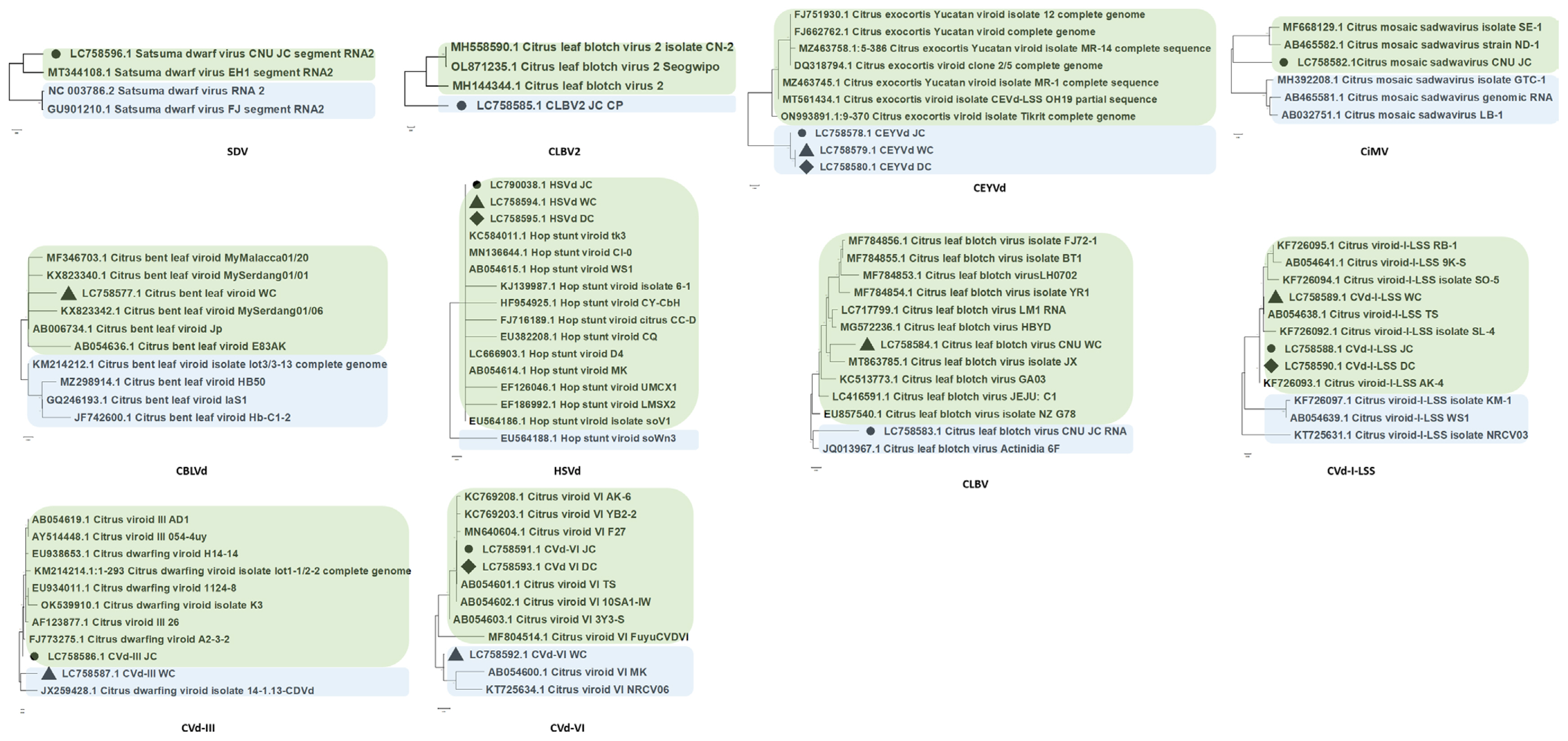
Phylogenetic analyses of identified viruses and viroids. The black circles indicate isolates identified from the Jeju library, the black triangles represent isolates from the Wando library, and the black diamonds depict isolates from the Dangjin library.
Analyses of SNPs for identified viruses and viroids
Viruses and viroids with RNA genomes exhibit high mutation rates, resulting in significant genetic diversity (Domingo and Holland, 1997; Gago et al., 2009). To investigate this diversity, SNP analysis was conducted for each virus and viroid (Fig. 6). SNPs were identified by aligning the raw reads to the reference genomes. In the Jeju isolates, CiMV (1,671 SNPs), SDV (1,093 SNPs), and CLBV2 (883 SNPs) exhibited the highest numbers of SNPs. In the Wando isolates, CLBV had the highest number of SNPs, with a total of 151. Viruses and viroids identified in Dangjin had a relatively lower number of SNPs compared to those from other regions, with CEYVd showing the highest count at 17, followed by only one SNP in CVd-I-LSS.
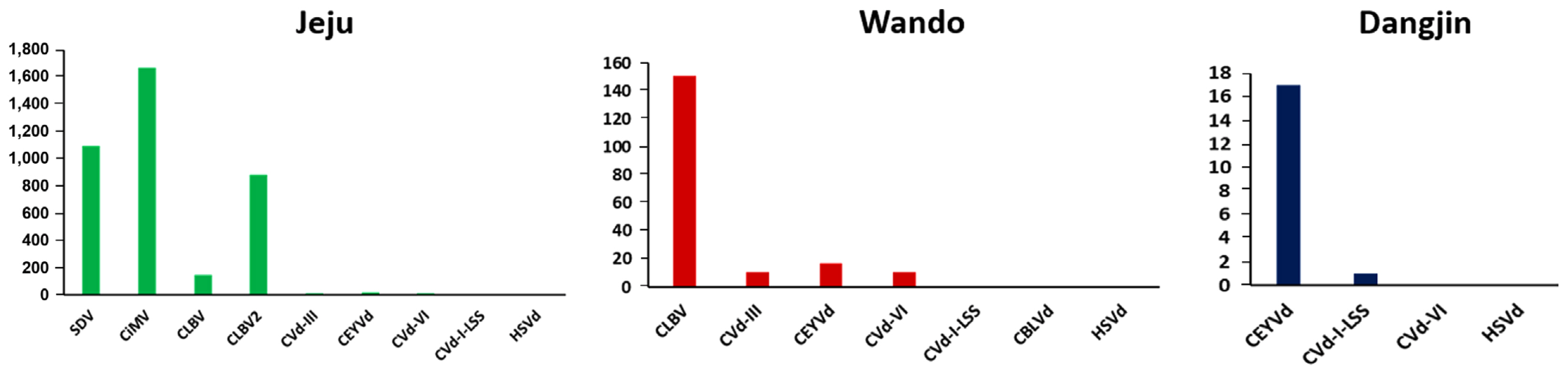
The number of identified single nucleotide polymorphisms for each virus or viroid. CBLVd, citrus bent leaf viroid; CEYVd, citrus exocortis Yucatan viroid; CiMV, citrus bent leaf viroid; CLBV, citrus leaf blotch virus; Cvd-I-LSS, citrus viroid-I-LSS; CVd-III, citrus viroid III; CVd-VI, citrus viroid VI; HSVd, hop stunt viroid; SDV, satsuma dwarf virus.
Validation of the identified viruses using RT-PCR
To validate the presence of the identified viruses and viroids, RT-PCR was conducted (Fig. 7). The RT-PCR was performed using primers specific to viruses and viroids that were either reported in previous studies or designed based on the consensus sequences (Table 2) (Astruc et al., 1996; Bernad and Durán-Vila, 2006; Cao et al., 2017; Hyun et al., 2020; Osman et al., 2015). For the viruses, the primers were designed based on the CP regions, while for the viroids, the primers were designed based on the complete sequences of the viroids. The expected amplicon size of all viruses identified in the three libraries through RNA-Seq was confirmed via RT-PCR. The citrus reference actin gene was also amplified. The amplicons were sequenced by Sanger sequencing, and the presence of each virus was confirmed in the obtained sequences through a BLASTN search (data not shown).
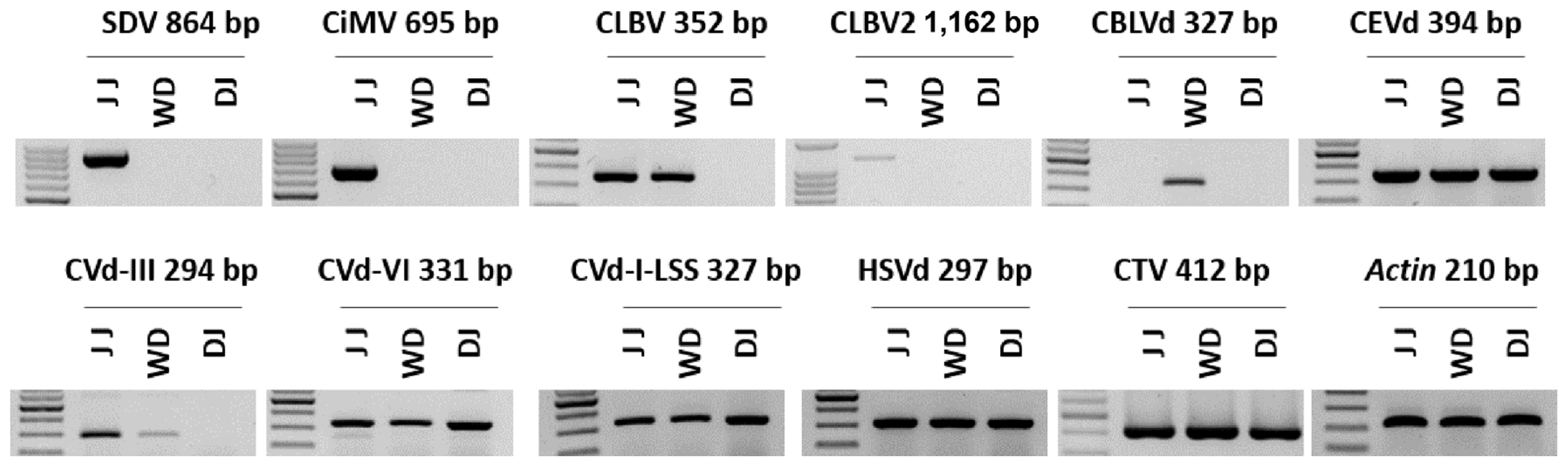
Confirmation of the identified viruses from each citrus transcriptome by RT-PCR. The amplified PCR products were visualized by 1.2% agarose gel electrophoresis. CBLVd, citrus bent leaf viroid; CEVd, citrus exocortis viroid; CiMV, citrus bent leaf viroid; CLBV, citrus leaf blotch virus; CTV, citrus tristeza virus; Cvd-I-LSS, citrus viroid-I-LSS; CVd-III, citrus viroid III; CVd-VI, citrus viroid VI; DJ, Dangjin; HSVd, hop stunt viroid; JJ, Jeju; SDV, satsuma dwarf virus; WD, Wando.
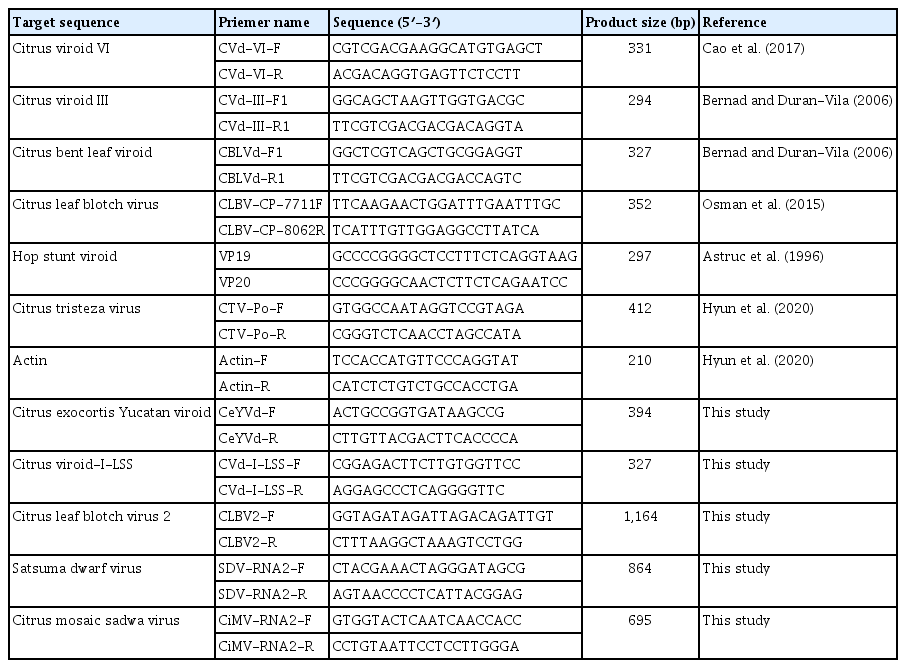
Sequences of primers used in this study
Discussion
Citrus cultivation, particularly on Jeju Island, stands as a significant agricultural industry in Korea, contributing substantially to the economy. Despite the global demand for citrus fruits highlighting their economic importance, the industry grapples with significant challenges due to viral diseases affecting citrus crops. As mentioned in the introduction, various major citrus viruses, including CTV and CEVd, have been identified as significant threats to citrus trees worldwide (Belabess et al., 2021; Roistacher and Moreno, 1991). Consequently, the quality and yield of citrus crops remain at risk due to these viral diseases.
Conventional diagnostic methods, primarily PCR-based nucleic acid amplification techniques, have been instrumental in pinpointing specific viruses. However, these methods have limitations, especially in simultaneously detecting multiple viruses and identifying unknown strains. To overcome these constraints, this study adopted HTS as a potent tool for identifying and characterizing viruses affecting Satsuma mandarins in Korea. Our study aimed to utilize HTS to identify and characterize viruses in Satsuma mandarins, one of Korea’s most popular citrus cultivars, and to explore the virosphere in major citrus cultivation areas.
The results provide a comprehensive overview of the viral landscape, unveiling the presence of diverse viruses and viroids in different regions (Jeju, Wando, and Dangjin). A key observation was the substantial variation in the types and quantities of viruses across these regions. Notably, CLBV predominated in Jeju, while CEVd and CVd-I-LSS were prevalent in Wando and Dangjin, respectively. Phylogenetic analysis further revealed the evolutionary relationships, with most viruses showing close relationships to isolates from China or Japan. A noteworthy contribution of this study is that, to our knowledge, it is the first report on the occurrence of CVd-I in Citrus spp. in Korea, as no previous cases have been documented. However, a point of concern emerged regarding the identification of CTV in all regions due to low accuracy in genome assembly. This challenge may stem from the relatively long RNA genome (approximately 19.3 kb) of CTV. Biases introduced during sample amplification in the library preparation process and insufficient overlap between fragments in short-read sequencing could account for this discrepancy (Boers et al., 2019). Additionally, the existence of at least eight CTV strains, combined with the pooling of samples, might have resulted in a mixture of different CTV strains (Garnsey et al., 2005). Consequently, achieving accurate mapping for a specific strain could have been compromised.
CTV is a highly destructive virus known to induce two diseases in citrus trees, leading to either the rapid decline of the tree or the formation of holes in the stem, depending on the host’s susceptibility or the combination of rootstock. Cross protection, a phenomenon that prevents secondary infection by the same or closely related viruses, has been commercially exploited with CTV being one of the first cases (Folimonova et al., 2020). However, its effectiveness has been limited to specific regions or certain varieties and has not been universally applicable. Current research is concentrated on identifying CTV isolates suitable for cross protection, and understanding the characteristics of CTV sources within citrus production regions is particularly crucial (Kleynhans and Pietersen, 2016; Lubbe, 2015). Hence, this study holds the potential to contribute significantly to these ongoing efforts.
In all three regions, the highly structured single-stranded RNA plant pathogens, CEVd and HSVd, were commonly detected (Flores et al., 2009). Of the seven known citrus viroids, only CEVd and HSVd have been reported to be associated with economically significant diseases in citrus, causing symptoms such as exocortis or cachexia, respectively (Belabess et al., 2021). Their presence is documented in almost all citrus growing regions worldwide, consistent with their detection in all the tested regions in Korea in our findings. These results underscore the importance of developing diagnostic methods and management strategies for CEVd and HSVd, significant viroids associated with citrus.
FPKM serves as a method for quantifying gene expression levels in RNA-Seq data, standardizing expression by accounting for both gene length and the number of sequenced reads. Specifically, the FPKM represents the relative amount of gene expression, enabling gene expression comparison across different samples (Jo et al., 2018; Zhao et al., 2021). In this study, the FPKM was employed to measure the quantity of viral and viroid RNA in citrus, specifically Satsuma mandarins, across diverse regions. Notably, Jeju demonstrates a prevalence of CLBV, whereas Wando and Dangjin exhibit higher expression levels of CEVd and CVd-I-LSS. Dangjin, in particular, was dominated by CEVd, thus emphasizing regional virus diversity. These findings offer insights into the relative expression levels of viruses in each region.
The genetic diversity revealed through SNP analysis underscores the high mutation rates of viruses with RNA genomes. For instance, in the Jeju region, CiMV (1,671 SNPs), SDV (1,093 SNPs), and CLBV2 (883 SNPs) exhibited the highest proportions of SNPs. In the Wando-gun, CLBV had the highest number of SNPs with 151. The viruses and viroids identified in the Dangjin-si showed relatively lower SNP counts, with CEVd having the highest at 17. This SNP analysis demonstrates the elevated mutation rates of viruses and viroids with RNA genomes, implying their potential role in adaptability and persistence within the citrus population.
We analyzed each regions’s citrus library to ascertain the presence of novel viruses. Employing Geneious software, we assembled individual reads to produce contigs and conducted MEGABLAST searches against the NCBI plant virus database for identifying viruses and viroids. Subsequently, contigs associated with identified viruses and viroids underwent BLASTx searches to identify viruses displaying high amino acid sequence similarity, potentially unveiling novel viruses. Regrettably, the analysis did not disclose any new viruses, confirming only the presence of identified viruses and viroids in this study.
While this study provides valuable insights, it is crucial to acknowledge its limitations. The exclusive focus on Satsuma mandarins and specific regions may not completely capture the broader spectrum of citrus viruses in Korea. Future study should consider expanding the scope to conduct a more comprehensive national survey. In conclusion, this study utilized HTS to unravel the intricate virosphere affecting Satsuma mandarins in Korea. The findings significantly contribute to ongoing efforts aimed at developing more effective strategies for preventing and controlling viral diseases in citrus crops.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This work was carried out with the support of Cooperative Research Program for Agriculture Science and Technology Development (Project No. PJ014947032023) Rural Development Administration, Republic of Korea.