A Revision of the Phylogeny of Helicotylenchus Steiner, 1945 (Tylenchida: Hoplolaimidae) as Inferred from Ribosomal and Mitochondrial DNA
Article information

Abstract
Identification of Helicotylenchus species is very challenging due to phenotypic plasticity and existence of cryptic species complexes. Recently, the use of rDNA barcodes has proven to be useful for identification of Helicotylenchus. Molecular markers are a quick diagnostic tool and are crucial for discriminating related species and resolving cryptic species complexes within this speciose genus. However, DNA barcoding is not an error-free approach. The public databases appear to be marred by incorrect sequences, arising from sequencing errors, mislabeling, and misidentifications. Herein, we provide a comprehensive analysis of the newly obtained, and published DNA sequences of Helicotylenchus, revealing the potential faults in the available DNA barcodes. A total of 97 sequences (25 nearly full-length 18S-rRNA, 12 partial 28S-rRNA, 16 partial internal transcribed spacer [ITS]-rRNA, and 44 partial cytochrome c oxidase subunit I [COI] gene sequences) were newly obtained in the present study. Phylogenetic relationships between species are given as inferred from the analyses of 103 sequences of 18S-rRNA, 469 sequences of 28S-rRNA, 183 sequences of ITS-rRNA, and 63 sequences of COI. Remarks on suggested corrections of published accessions in GenBank database are given. Additionally, COI gene sequences of H. dihystera, H. asiaticus and the contentious H. microlobus are provided herein for the first time. Similar to rDNA gene analyses, the COI sequences support the genetic distinctness and validity of H. microlobus. DNA barcodes from type material are needed for resolving the taxonomic status of the unresolved taxonomic groups within the genus.
Spiral nematodes of the genus Helicotylenchus are some of the most common plant-parasitic nematodes occurring in various natural and agricultural ecosystems. The genus is cosmopolitan and comprises more than 230 nominal species described worldwide (Marais, 2001; Mwamula et al., 2020b; Subbotin et al., 2011). Many of the described species are normally part of the root microbiome associated with rhizospheric soils of various crops of agricultural importance though with no reported substantial damage on host crops (Subbotin et al., 2011). However, several species have been implicated in severe economic damage on some agricultural crops and grasses. For instance, H. multicinctus (Cobb, 1893) Golden 1956, H. dihystera (Cobb, 1893) Sher 1961, H. variocaudatus (Luc, 1960) Fortuner, 1984, and H. erythrinae (Zimmermann, 1904) Golden, 1956 are serious pathogens on banana (De Waele and Elsen, 2007; Riascos-Ortiz et al., 2020; Van Den Berg et al., 2003); H. pseudorobustus (Steiner, 1914) Golden 1956, H. digonicus Perry et al., 1959, H. indicus Siddiqi, 1963, H. cavenessi Sher, 1966, H. microcephalus Sher, 1966, H. oleae Inserra et al., 1979, and H. microlobus Perry et al., 1959 have been reported to cause damage to various agricultural crops and turfgrass (Inserra et al., 1979; Mwamula and Lee, 2021; O’Bannon and Inserra, 1989; Subbotin et al., 2011; Zeng et al., 2012).
Helicotylenchus species are a taxonomically confounding group. Apart from species that have unique morphological characters such as H. multicinctus, the identification of other species is very challenging due to phenotypic plasticity within the key diagnostic characters (Subbotin et al., 2011, 2015). As a result, species boundaries are not well defined. This is even exacerbated by the existence of cryptic species within the genus, and possible existence of mixed species populations in common habitats. Even among the most reported species such as H. pseudorobustus, H. dihystera, H. digonicus, H. microlobus, and H. vulgaris Yuen, 1964, misidentifications are evident in published literature due to under- or over-estimation of intra- and interspecific phenotypic variability of various morphological characters during diagnosis. Also of interest is the continuous lack of consensus among taxonomists on the validity of some species. The taxonomic position of H. microlobus in relation to H. pseudorobustus is still controversial following protracted contestation of the validity of the former (Divsalar et al., 2020; Fortuner, 1984; Fortuner et al., 1981, 2018; Mwamula et al., 2020b; Subbotin et al., 2011, 2015).
DNA barcoding is a powerful tool for nematode identification. In recent years, inferences from ribosomal DNA have been used for discriminating cryptic, and (or) very closely related species and reconstruction of phylogenetic relationships within Helicotylenchus (Mohammadi Zameleh et al., 2020; Mwamula et al., 2020b; Shokoohi et al., 2021; Subbotin et al., 2011, 2015). Specifically, attempts to discriminate a population of H. pseudorobustus recovered from type locality from many populations of related species, including H. microlobus were made by Subbotin et al. (2015). Despite their demonstration of the taxonomic status of H. microlobus as a valid species based on the two commonly used barcodes (internal transcribed spacer [ITS]-rRNA and 28S-rRNA) for the genus, consensus is yet to be reached among scholars. A combination of ribosomal DNA (ITS-rRNA, 28S-rRNA) and mitochondrial DNA has been shown to offer precision in species identification among various nematode groups (Mwamula et al., 2020a; Subbotin et al., 2018; Vovlas et al., 2015). Except for a few species, Helicotylenchus species have been molecularly characterized using primarily ribosomal DNA fragments. Mitochondrial DNA (cytochrome c oxidase subunit I [COI] gene) has only been reported in a limited number of species, including H. oleae, H. canadensis, H. pseudorobustus, H. varicaudatus, and H. vulgaris (Palomares-Rius et al., 2018; Rybarczyk-Mydłowska et al., 2019). Therefore, the objectives of this study were as follows: (1) to characterize H. microlobus and other common Helicotylenchus species occurring in turfgrass ecosystems in Korea using COI gene and make a comparison with the available COI sequences of H. pseudorobustus; (2) review the phylogenetic relationships within all published Helicotylenchus species using ribosomal (18S-, 28S-, and ITS-rRNA) and mitochondrial DNA (COI gene) as inferred from Bayesian phylogenetic inference approach.
Materials and Methods
Nematode populations and extraction
The various Helicotylenchus spp. populations were recovered from rhizospheric soil samples collected from 9 golf courses. Detailed information regarding the sample localities, host plant, and laboratory codes is summarized in Table 1. The nematodes were extracted from sod samples using a combination of modified Cobb’s sieving and Baermann funnel methods (Whitehead and Hemming, 1965). With the aid of a Nikon SMZ 1000 stereomicroscope (Nikon, Tokyo, Japan), nematode specimens belonging to Helicotylenchus spp. were picked out from the collected nematode suspension and subsequently characterized to species level based on inferences from morphometrics and DNA sequence data.
Locality of each Helicotylenchus species population used in this study
Morphological characterization
A defined number of specimens from each population were heat-killed, fixed, and mounted to pure glycerin (Seinhorst, 1959). Morphometric data were taken using a Zeiss imager Z2 microscope (Carl Zeiss, Jena, Germany) fitted with Axio-vision software Material Science Software for Research and Engineering (Carl Zeiss). Morphological species identification was done following the identification keys and species descriptions compiled by Boag and Jairajpuri (1985), Fortuner (1984), Marais (2001), Mwamula et al. (2020b), and Subbotin et al. (2015).
Molecular characterization
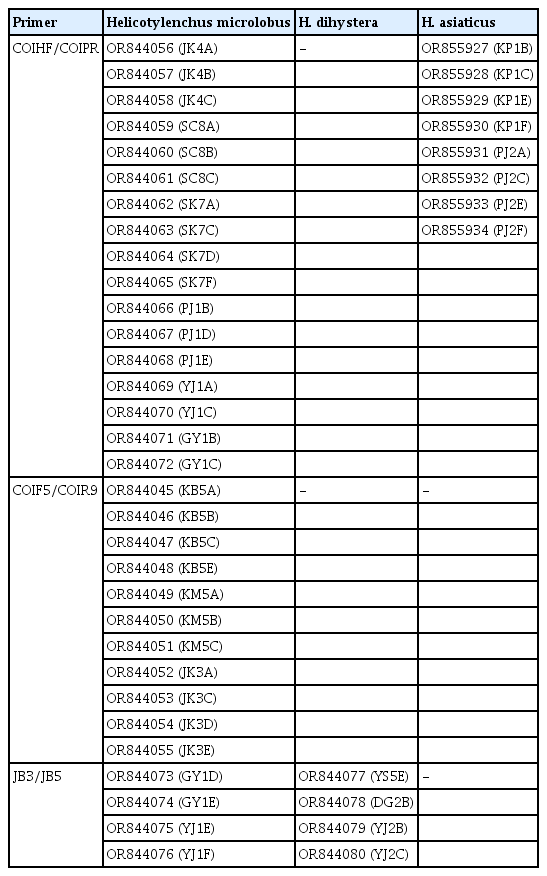
Ribosomal and mitochondrial DNA was extracted from single female and male specimens using the DNA extraction kit WizPure, as described by Iwahori et al. (2000). The 18S-rRNA and COI gene of the previously published Helicotylenchus populations (Mwamula et al., 2020b) were amplified from their stored DNA. The nearly full-length 18S-rRNA gene was amplified as 2 partially overlapping fragments using 2 primer sets: 988F (5′-CTCAAAGATTAAGCCATGC-3′) and 1912R (5′-TTTACGGTCAGAACTAGGG-3′), 1813F (5′-CTGCGTGAGAGGTGAAAT-3′), and 2646R (5′-GCTACCTTGTTACGACTTTT-3′) (Holterman et al., 2006); D2A (5′-ACAAGTACCGTGAGGGAAAGTTG-3′) and D3B (5′-TCGGAAGGAACCAGCTACTA-3′) (Nunn, 1992) amplified the D2-D3 expansion segment of 28S-rRNA; TW81 (5′-GTTTCCGTAGGTGAACCTGC-3′) and AB28 (5′-ATATGCTTAAGTTCAGCGGGT-3′) (Curran et al., 1994), and S-ITS1 (5′-TTGATTACGTCCCTGCCCTTT-3′) and Vrain2R (5′-TTTCACTCGCCGTTACTAAGGGAATC-3′) (Vrain et al., 1992) amplified the partial ITS-rRNA region. The partial COI gene was amplified using a combination of several primer sets: COIF5/COIR9; JB3/JB5 and COIHF/COIPR as detailed in Table 2. Polymerase chain reaction (PCR) was performed with a PCR cycler (T100, Bio-Rad, Hercules, CA, USA), the PCR program with specific primers being set as follows: initial denaturation at 95°C for 5 min, 35 cycles at 95°C for 30 s, followed by an annealing step at 58°C for 30 s (S-ITS1/Vrain2R), 54°C for 40 s (COIHF/COIPR), 56°C for 40 s (COIF5/COIR9); 72°C for 60 s (S-ITS1/Vrain2R), 72°C for 80 s (COIHF/COIPR and COIF5/COIR9), and finally one cycle at 72°C for 5 min (COIHF/COIPR) and 72°C for 10 min (S-ITS1/Vrain2R and COIF5/COIR9). The cycle for D2A/D3B and JB3/JB5 primer sets was as described by Mwamula et al. (2023a) while the thermal profiles for TW81/AB28, 988F/1912R and 1813F/2646R primer sets were set as detailed by Mwamula et al. (2023b). The PCR products were purified using a PCR purification kit (Qiagen, Hilden, Germany) and quantified using a quickdrop spectrophotometer (Molecular Devices, San Jose, CA, USA). The purified products were subsequently used for direct sequencing in both directions using the same primers as specified above. DNA sequencing was performed at Macrogen. The edited DNA sequences were submitted to the GenBank database under the accession nos.: OR843993-OR844017 (18S-rRNA); OR844019-OR844030 (28S-rRNA); OR852617-OR852632 (ITS-rRNA); OR844045-OR844080 and OR855927-OR855934 (COI gene).

Overview of PCR primers used for amplification of the partial COI gene
Phylogenetic analysis
The obtained and edited sequences (18S-rRNA, 28S-rRNA, ITS-rRNA and COI gene) were aligned using ClustalX (Thompson et al., 1997) along with the sequence data sets of Helicotylenchus species published in GenBank (Divsalar et al., 2020; Ehtesham et al., 2021; Etongwe et al., 2020; Mohammadi Zameleh et al., 2020; Mwamula et al., 2020b; Palomares-Rius et al., 2018; Riascos-Ortiz et al., 2020; Rybarczyk-Mydłowska et al., 2019; Shokoohi et al., 2018, 2021; Subbotin et al., 2011, 2015; Xia et al., 2022). Only a representative number of 18S-rRNA sequences were included in the data set for analysis. The data sets for 28S-rRNA, ITS-rRNA, and COI gene comprised essentially all the comparable sequences of Helicotylenchus species available in GenBank database at the time of the current analysis. However, the too divergent sequences possibly belonging to different genera or deemed to contain errors were omitted from phylogenetic reconstruction. The partial sequences of Rotylenchus urmiaensis (KP718969) and Scutellonema bradys (AJ966504) were selected as the outgroup taxa for the 18S-rRNA gene; Hoplolaimus galeatus (EU626787) and Hoplolaimus seinhorsti (DQ328752) for 28S-rRNA; Scutellonema bradys (JX472067) and Rotylenchus pumilus (JX015436) for ITS-rRNA; and Bursaphelenchus mucronatus (AB067765) and Bursaphelenchus sinensis (AB232163) for COI gene. The generated alignments were analyzed with Bayesian inference (BI) using MrBayes 3.2.7 (Ronquist et al., 2012) under the GTR + I + G model. BI analysis for each gene was initiated with a random starting tree and run with four chains for 1 × 106 generations. The Markov chains were sampled at intervals of 100 generations. After discarding burn-in samples, a consensus tree was generated with a 50% majority rule. Trees were subsequently visualized and edited using FigTree v1.4.4 software. Posterior probabilities (PP) exceeding 50% are given on appropriate clades. Interspecific and intraspecific sequence variations were examined using PAUP* v4.0a169 (Swofford, 2003).
Results
Species identification and delimitation
Identification of the newly recovered populations was made using an integrative approach considering morphological characteristics and the results of sequence analyses and phylogenetic inferences. A total of 97 sequences (25 nearly full-length 18S-rRNA, 12 partial 28S-rRNA, 16 partial ITS-rRNA, and 44 partial COI gene sequences) were newly obtained from the various populations of the four studied species in the present study. All the newly recovered populations were identified morphologically as representatives of the same species published by Mwamula et al. (2020b). The morphological and morphometric data of the current populations of H. microlobus agree well with those of Mwamula et al. (2020b), Siddiqi (1972, 2000), Subbotin et al. (2015), and Tzortzakakis et al. (2018). Helicotylenchus dihystera population YJ2 and H. caudatus Sultan 1985 population PJ3 are morphologically and morphometrically similar to the topotypes detailed by Sher (1966) and Sultan (1985), respectively. Helicotylenchus asiaticus is herein recorded for the first time outside its type locality. The morphometric data for the newly recovered populations are detailed in Table 3. The partial COI gene sequences of the studied species (H. microlobus, H. dihystera, and H. asiaticus) are herein supplied for the first time. Similarly, the partial 18S-rRNA gene sequences of H. caudatus and H. asiaticus are also reported for the first time.

Morphometrics of Helicotylenchus spp. populations from turfgrass in Korea
Molecular characterization and phylogenetic relationships
The 18S-rRNA gene
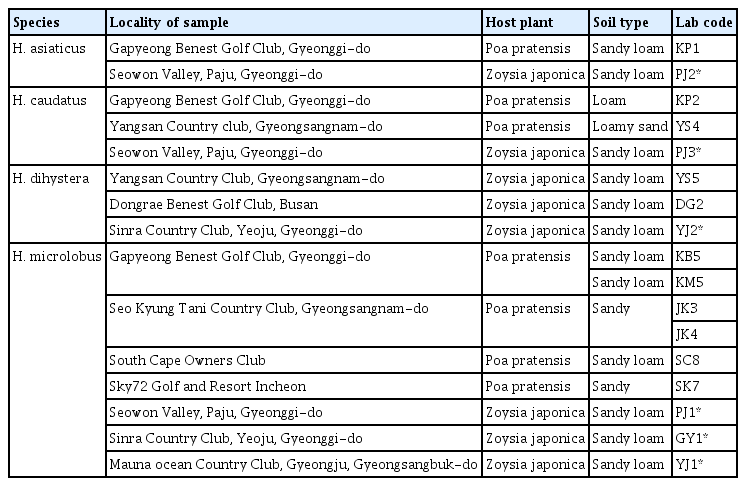
The amplification of the nearly full-length 18S-rRNA gene from all the studied species and their respective populations yielded fragments of approximately 1,650–1,700 bp. The partial 18S-rRNA gene sequence alignment was 1,685 bp in length and contained 103 sequences of Helicotylenchus and 2 sequences of the outgroup taxa (Scutellonema bradys and Rotylenchus urmiaensis). Phylogenetic relationships between Helicotylenchus species, as inferred from Bayesian phylogenetic analysis of the dataset are shown in Fig. 1. Seven moderately supported major clades were distinguished within the analyzed taxa. Sequence variation within all taxa including outgroups reached 114 bp (6.8%) and for Helicotylenchus, it reached 91 bp (5.4%). Minimal to moderate sequence variation (0–45 bp [0.0–2.7%]) was evident among taxa belonging to Clade I (PP = 84%). The clade comprised eleven putative taxa including H. caudatus, H. indicus, H. multicinctus, H. digitiformis, H. paraplatyurus, H. pseudorobustus, H. certus, H. microlobus, H. crenacauda, H. dihystera, H. rotundicauda and two unidentified populations (Helicotylenchus sp. 1 JH-2014 and Helicotylenchus sp. 5 JH-2014).
Bayesian tree inferred under the GTR + I + G model from 18S-rRNA gene sequences of Helicotylenchus species. Posterior probability values exceeding 50% are given on appropriate clades. The studied populations are indicated in bold dark blue text. Outgroup taxa: Rotylenchus urmiaensis and Scutellonema bradys.
All four newly obtained sequences of H. caudatus were identical with no recorded intraspecific variation and differed from the closely clustered H. indicus sequences by 23–26 bp (1.4–1.6%). Intraspecific sequence variation for the newly obtained H. microlobus sequences varied by 0–2 bp and these differed from H. microlobus populations from China; OP225352 and ON117514 by 0–9 bp (0.0–0.6%) and from the closely clustered taxa (H. digitiformis, H. crenacauda, H. pseudorobustus, H. certus, Helicotylenchus sp. 5 JH-2014, Helicotylenchus sp. 1 JH-2014, including H. dihystera accessions: KJ934127 and JX069950) by 0–26 bp (0.0–1.6%). An intraspecific sequence variation of 0–2 bp was recorded among the obtained H. dihystera sequences. The new sequences differed from other H. dihystera populations by 0–24 bp (0.0–1.5%) and from the closely clustered H. rotundicauda (KM603516 and OP225358) by 2–8 bp. Helicotylenchus asiaticus sequences formed a moderately supported independent clade (PP = 87%), with no recorded intraspecific sequence variation.
The 28S-rRNA gene
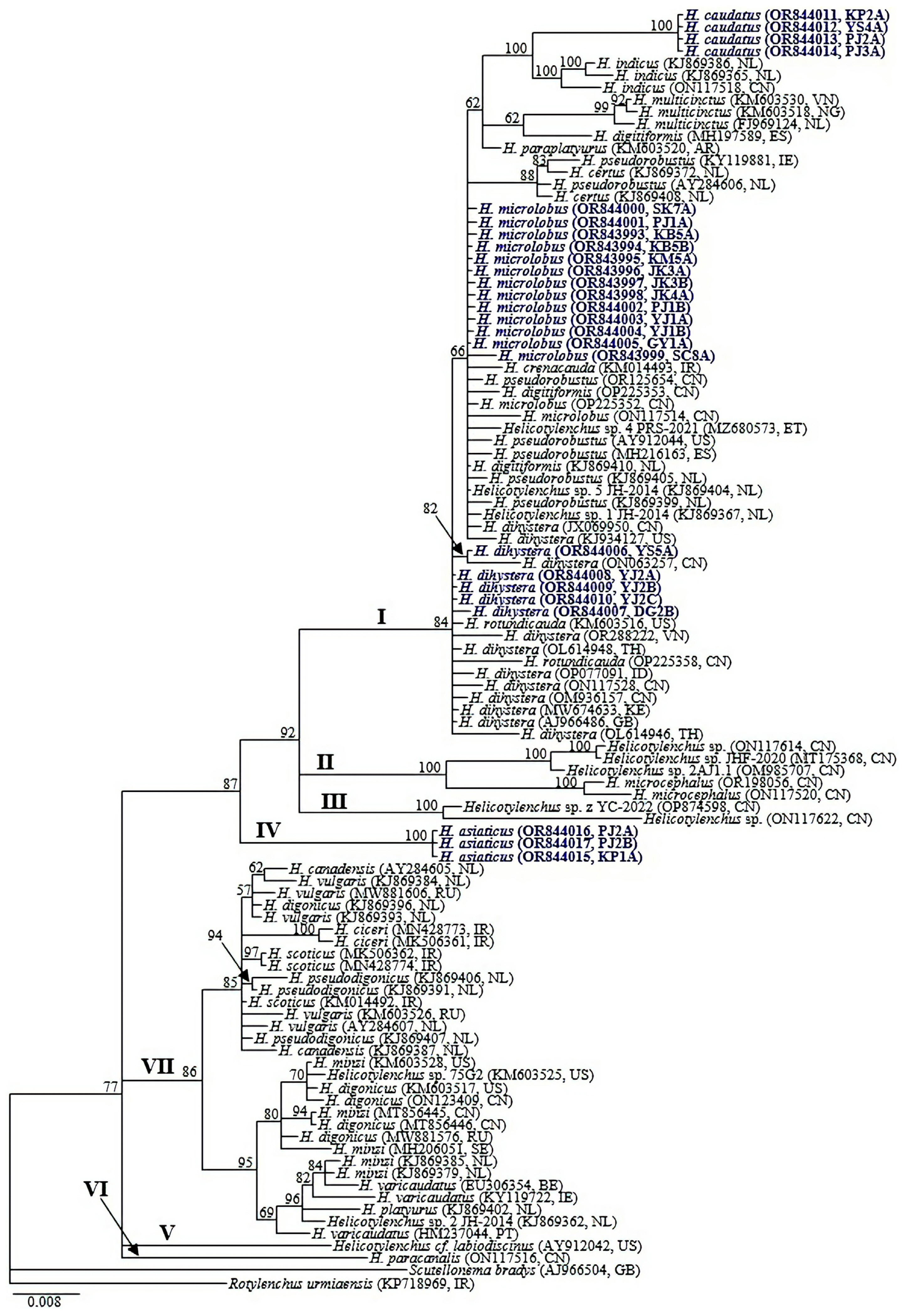
The amplicons of the D2–D3 expansion segment of 28S-rRNA gene were approximately 750 bp in length. The partial 28S-rRNA gene sequence alignment constituted all the comparable sequences of Helicotylenchus published in GenBank at the time of this analysis. A few incomparable sequences were excluded from the analysis (see remarks on omitted sequences). The alignment contained 471 sequences of Helicotylenchus including 2 sequences of the outgroup taxa (Hoplolaimus galeatus and Hoplolaimus seinhorsti). Twelve highly and moderately supported clades were distinguished within the taxa. Phylogenetic relationships between Helicotylenchus species, as inferred from Bayesian phylogenetic analysis of the dataset are shown in Fig. 2A–E. The general topology of the resultant Bayesian tree agrees with the phylogenetic data presented by Subbotin et al. (2015). Intraspecific sequence variation for the three newly obtained sequences of H. microlobus was 0–1 bp, and the intraspecific sequence variation for all H. microlobus sequences varied from 0–10 bp (0.0–1.8%); for H. pseudorobustus − 0–11 bp (0.0–2.0%); and for H. paxilli − 0–5 bp (0.0–0.8%). Interspecific sequence diversity within H. microlobus, H. paxilli, and H. pseudorobustus sequences reached 29 bp (6.0%) (15–29 bp [2.6–6.0%]). Intraspecific sequence variation for H. digonicus “Type A” varied from 0–15 bp (0.0–2.6%); and for H. broadbalkiensis − 1–6 bp (0.1–1.0%). Sequence variation within all comparable H. dihystera sequences (except ON117414) varied from 0–15 bp (0.0–2.6%). High sequence divergences of 11–21 bp (1.9–3.6%) were recorded in pairwise comparison of ON117414 with all other H. dihystera accessions. Intraspecific sequence variation for H. paraplatyurus including Helicotylenchus sp. X-1 (KM506841-KM506843) varied by 0–3 bp (0.0–0.5%) and for H. californicus − 0–1 bp. Sequences of H. caudatus from Korea (including the newly obtained sequences) varied by 1–6 bp (0.1–0.8%) but differed from the shorter sequences of H. caudatus from Ethiopia (OP647413-OP647416) by 12–16 (2.7–3.7%). Intraspecific sequence variation within H. asiaticus was 0–2 bp and these differed from the closely clustered unidentified species from Korea (KY484829-KY484832) by 5–9 bp (0.6–1.2%). Sequence diversity within H. multicinctus varied by 0–13 bp (0.0–2.5%); for H. labiodiscinus − 3–9 bp (0.5–1.5%); for H. oleae − 0–8 bp (0.0–1.1%); for H. cuspicaudatus − 3–12 bp (0.3–1.8%); for H. microcephalus including Helicotylenchus sp. VII − 0–6 bp (0.0–0.8%); for H. digonicus “Type B” and H. minzi − 0–13 bp (0.0–2.2%); and for H. vulgaris Type A and H. canadensis − 0–6 bp (0.0–1.0%).
(A–E) Bayesian tree inferred under the GTR + I + G model from LSU D2–D3 partial sequences of Helicotylenchus species. Posterior probability values exceeding 50% are given on appropriate clades. The studied populations are indicated in bold dark blue text. Outgroup taxa: Hoplolaimus galeatus and Hoplolaimus seinhorsti.
The ITS-rRNA gene
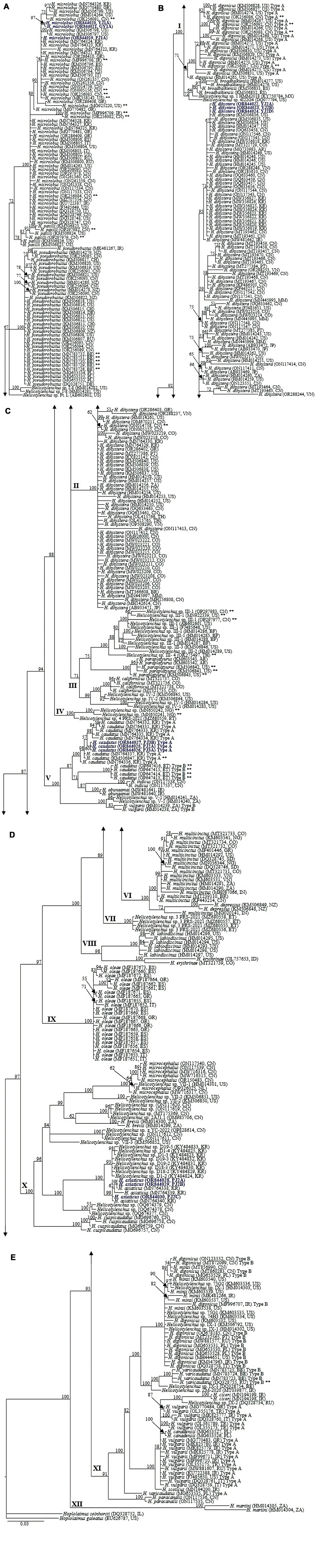
The amplification of the partial ITS-rRNA gene yielded single amplicons of approximately 1,200 bp. The ITS-rRNA gene sequence alignment contained 185 sequences of Helicotylenchus including 2 sequences of outgroup taxa (Scutellonema bradys and Rotylenchus pumilus). Phylogenetic relationships between Helicotylenchus species, as inferred from Bayesian phylogenetic analysis of the dataset are shown in Fig. 3A–C. Ten highly and moderately supported clades were distinguished within the taxa. ITS-rRNA gene sequence variation for H. microlobus populations varied from 0–48 bp (0.0–4.9%); for H. digonicus − 4–16 bp (0.4–1.6%), differing from the closely clustered H. ciceri (MN194203 and MN194204) by 85–96 bp (8.8–9.8%); for H. broadbalkiensis − 12–14 bp (1.2–1.5%), differing from H. scoticus (MN194205) by 40–50 bp (4.0–5.0%); for H. pseudorobustus sensu stricto populations − 0–5 bp (0.0–0.6%); and for H. paxilli − 1–15 bp (0.1–1.5%). Sequence diversity within all but four (ON123797-ON123799 and MH142618) H. dihystera sequences varied from 0–33 bp (0.0–3.2%). The accessions ON123797-ON123799 and MH142618 contain a significant number of nucleotide deletions, insertions, and substitutions which might have been caused by sequencing errors. Pairwise comparison with other H. dihystera sequences showed sequence variation of 21–59 bp (2.5–5.4%). Despite the disparities, these accessions clustered together with other H. dihystera sequences (Fig. 3A). The top BLASTN hit for the H. caudatus ITS-rRNA gene sequences was H. crenacauda (DQ309586) with a percentage identity of 85% and 100% query coverage. The newly obtained H. caudatus sequences varied by 0–2 bp and differed from the closely clustered H. abunaamai (MW488036, MW488037), H. crenacauda (DQ309586), H. caudatus from Ethiopia (OP650246) and H. indicus (ON128730, ON128731), by 29–59 (7.2–9.2%), 92–158 bp (11.4–13.8%), 93–130 (14.9–15.6%), and 114–158 (16.6–19.5%), respectively. Intraspecific sequence variation within H. multicinctus varied from 1–37 bp (0.08–3.0%); for H. oleae − 5–24 bp (0.9–3.0%); for H. cuspicaudatus − 1–29 bp (0.07–2.3%); for H. microcephalus − 0–26 bp (0.0–2.3%); and for H. asiaticus − 0–8 bp (0.0–0.7%), differing from closely clustered Helicotylenchus spp. (KY484812-KY484814, KY484818-KY484822, KY512778, KY512779, and OR159472) by 42–56 bp (4.3–5.2%).
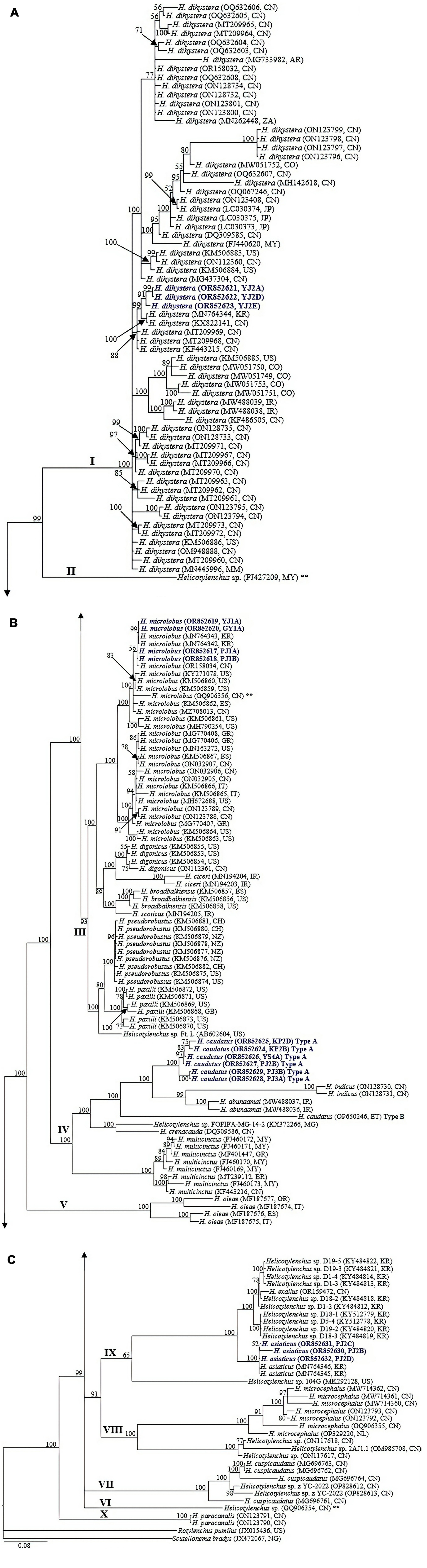
(A–C) Bayesian tree inferred under the GTR + I + G model from ITS-rRNA gene sequences of Helicotylenchus species. Posterior probability values exceeding 50% are given on appropriate clades. The studied populations are indicated in bold dark blue text. Outgroup taxa: Scutellonema bradys and Rotylenchus pumilus.
The COI gene
Amplification of the partial CO1 gene using the primer sets: COIF5/COIR9, COIHF/COIPR, and JB3/JB5 yielded single amplicons of ca 700, 400, and 350 bp in length, respectively. By using the primer set COIHF/COIPR, we were able to easily obtain amplicons for all four species. However, due to limited DNA template for H. caudatus and H. dihystera, amplicon sequencing was only done for two species (H. microlobus and H. asiaticus) and their respective studied populations. JB3/JB5 successfully amplified the target region for two (H. microlobus and H. dihystera) of the four species while COIF5/COIR9 was successful for only H. microlobus (Table 4). We also included the recently designed primer set M2F (5′-ATTGGiGSTTTTGGTAATT-3′) and RH1R (5′-CCAACAATGAATATATGATG-3′) (Rybarczyk-Mydłowska et al., 2019). However, amplification of the target region was not successful despite M2F/RH1R being the most successful primer set in the molecular studies performed by Rybarczyk-Mydłowska et al. (2019). The partial COI gene sequence alignment used in phylogenetic reconstruction was limited to 398 bp in length and contained 63 sequences of Helicotylenchus and two sequences of Bursaphelenchus used as outgroup taxa. Relationships between Helicotylenchus species, as inferred from Bayesian phylogenetic analysis of the dataset are shown in Fig. 4. Pairwise sequence comparison between the contentious H. microlobus and H. pseudorobustus was done using a longer alignment of 691 bp. Helicotylenchus microlobus differed from H. pseudorobustus by 13.4% (84 bp of the 628 aligned nucleotides) to 13.7% (64 bp of the 465 aligned nucleotides). Intraspecific sequence variation within all the newly obtained sequences of H. microlobus varied by 0–3 bp. Helicotylenchus microlobus and H. pseudorobustus clustered together with the new sequences of H. dihystera in a well-supported clade (PP = 100%). Intraspecific sequence variation within H. dihystera varied by only 0–1 bp. The newly obtained sequences of H. asiaticus also varied by 0–1 bp, and clustered together with H. oleae, H. varicaudatus, H. canadensis, and H. digonicus.
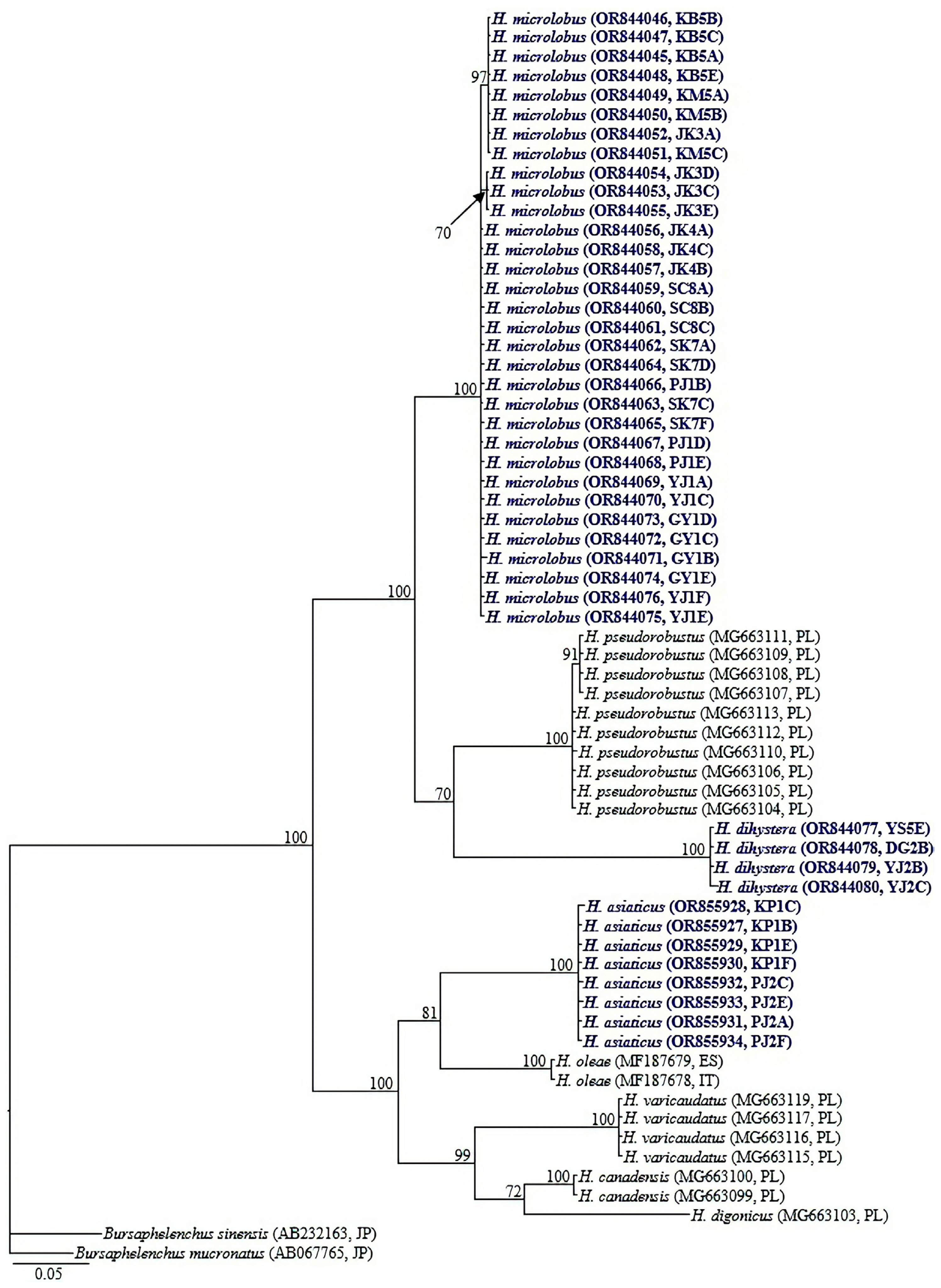
Bayesian tree inferred under the GTR + I + G model from cytochrome c oxidase subunit I (COI) gene sequences of Helicotylenchus species. Posterior probability values exceeding 50% are given on appropriate clades. The studied populations are indicated in bold dark blue text. Outgroup taxa: Bursaphelenchus mucronatus and Bursaphelenchus sinensis.
Suggested corrections of published accessions
The pairwise sequence comparisons and phylogenetic analyses revealed several possible misidentifications or mislabeling of several Helicotylenchus species in GenBank database. Suggested changes are indicated with double asterisk signs on the phylogenetic tree Figs. 2A–E and 3A–C. A comparison of the D2–D3 sequence data accessioned ON005156 and identified as H. dihystera with H. microlobus sequences revealed that it belongs to H. microlobus. Pairwise sequence comparison revealed intraspecific sequence variation of 0–9 bp (0.0–1.4%). The accession also significantly differed from all H. dihystera sequences by 17–30 bp (2.2–4.2%). Accessions MF996708, MF996709, MG925220, MK358143, and OK256081-OK256084 (all identified as H. pseudorobustus) clustered with H. microlobus. Sequence variation varied from 0–18 bp (0.0–3.1%); with the highest variation of 13–18 bp being recorded in sequence comparison with the short sequences accessioned MF996709 and OK256082. The accession OP297979 and OP297982 identified as H. microlobus and Helicotylenchus sp., respectively, probably belong to H. paxilli (differed from H. paxilli by 1–3 bp and 4–5 bp, respectively); OK256086-OK256088, and OK256092 identified as H. pseudorobustus probably belong to H. digonicus “type A” (differed from H. digonicus “type A” by 0–8 bp [0.0–1.4%]); MN783728-MN783732 (Helicotylenchus sp.) belong to H. pseudorobustus (differed from H. pseudorobustus by 0–10 bp [0.0–2.0%]); ON014758 (identified as H. microlobus) belong to H. dihystera (showed intraspecific variation with H. dihystera of 0–9 bp, and differed from H. microlobus by 18–33 bp [3.1–5.7%]); accession OK275497, identified as H. dihystera is herein designated as Helicotylenchus sp. III-4 based on the grouping of Subbotin et al. (2015). It differed from H. dihystera sequences by 22–34 bp (2.9–4.5%); OP297983 (identified as Helicotylenchus sp.), MN922339, and OP297977 (identified as H. dihystera) probably belong to Helicotylenchus sp. III-1 (intraspecific variation of 2–13 bp [0.3–2.2%] with Helicotylenchus sp. III-1 but differed from H. dihystera by 22–36 bp [2.9–4.8%]); KM506841-KM506843 (designated as Helicotylenchus sp. X-1 by Subbotin et al., 2015) belong to H. paraplatyurus (intraspecific variation of 1–3 bp); KM506847 (Helicotylenchus sp. V-2) belongs to H. caudatus (intraspecific variation of 0–4 bp); HM014301 (Helicotylenchus sp. V-II) belongs to the same taxa as H. microcephalus (intraspecific variation of 1–5 bp); and DQ328755 (Helicotylenchus sp. IX-3) belongs to the same taxa as H. varicaudatus “Type B” (MN783723-MN783725) (intraspecific variation of 3–5 bp).
Based on the ITS-rRNA gene sequence analysis, accession FJ427209 identified as H. dihystera from Malaysia probably represents unidentified species. It significantly differed from all other H. dihystera sequences by 53–101 bp (7.6–12.8%) and formed a separate independent clade from other H. dihystera accessions (Fig. 3A). Similarly, accession GQ906354 identified by Zhao et al. (2010) as H. dihystera is also too divergent from all Helicotylenchus spp. (up to over 200 bp, with 80% coverage) and formed an independent clade (Fig. 3C). Accession GQ906356 also identified by Zhao et al. (2010) as H. crenacauda most likely belongs to H. microlobus. The sequence evidently contains multiple nucleotide insertion and deletions (indels), including nucleotide substitutions. However, the accession clustered together with H. microlobus sequences and differed by 13–60 bp (1.4–6.7%). The alterations might be a result of sequencing errors, misassembles or presence of cloning vectors.
Remarks on omitted sequences
Some of the notable excluded sequences included the following accessions. For the 28S-rRNA gene, accessions MH087063, MH087064, MH084946, MH087062, and MF996772 (all identified as H. multicinctus from India) were omitted due to their significant divergence from all H. multicinctus sequences (differences of up to 200 bp) and all other Helicotylenchus spp. These accessions probably represent inaccurately edited sequence data. Accession MT321730 (H. dihystera) was omitted due to the existence of unusual multiple degenerate DNA letter codes. For the ITS-rRNA gene, MW404014 belongs to Merlinius sp. but was mistakenly labeled as H. digonicus (see Abdulsalam et al., 2021); GQ906351-GQ906353 identified by Zhao et al. (2010) as H. digonicus were also omitted. These accessions are highly divergent from all other Helicotylenchus spp. and are closer to Merlinius spp; accession MZ706973 identified as H. multicinctus from India is significantly divergent from H. multicinctus sequences, with biased substitution patterns and indels despite the 98% query coverage; ON117615 and ON117616 were omitted due to limited query coverage of below 30% when compared with other Helicotylenchus spp.; MW812365 and MN262451 identified by Abdulsalam et al. (2021) and Lamula (2020), respectively, as H. dihystera probably contain sequencing errors in form of multiple nucleotide insertion and deletions (indels), including multiple nucleotide substitutions and hence, when included in the analysis data, they generated a poor alignment that bore a substantial effect on phylogenetic reconstruction (data not shown).
Discussion
The use of molecular markers (ribosomal and mitochondrial DNA) in identification of Helicotylenchus is not only a quick and reliable nematode diagnostic tool but is crucial for discriminating morphologically related species and resolving cryptic species complexes within this speciose genus. The current analysis re-emphasizes the unsuitable nature of the 18S-rRNA gene in resolving phylogenetic relationships within closely related congeneric nematode species (Lazarova et al., 2019; Subbotin et al., 2008). Being a highly conserved gene compared to the 28S-rRNA, ITS-rRNA, and COI genes, the 18S gene is most useful for reconstructing deep nematode phylogeny at higher taxonomic levels (Blaxter et al., 1998; Holterman et al., 2009; van Megen et al., 2009). In the current analysis, only the more distant congeneric species such as H. caudatus, H. indicus, H. microcephalus, H. asiaticus, and H. paracanalis were distinguished from other related taxa based on the partial 18S-rRNA gene.
The D2–D3 region of the 28S-rRNA gene appears to be a comparatively reliable barcoding gene for the genus despite the unresolved taxonomic positions of H. digonicus “Type B” in relation to H. minzi, and H. vulgaris “Type A” with H. canadensis. The D2–D3 region is generally known to be more conserved within a species and does not suffer from high substitution saturation at a low taxonomic level compared to the ITS-rRNA gene (Bae et al., 2009; Subbotin et al., 2005). The unresolved taxonomic groups within the genus attests to the difficulties involved in distinguishing the morphologically close species, in addition to the possible existence of cryptic species complexes. For instance, according to Yuen (1964), H. vulgaris was differentiated from H. canadensis by the length of dorsal oesophageal gland orifice (9–12 vs. 8–10) and doubling (subdivisions) of the distal tail annuli. But these characters are known to be highly variable among Helicotylenchus spp. (Fortuner et al., 2018). In fact, the illustrations of the distal tail annuli of the population identified as H. vulgaris by Shokoohi et al. (2021) do not conform to the original illustrations of H. vulgaris but rather to that of H. canadensis (see Yuen, 1964; Waseem, 1961).
Helicotylenchus digonicus and H. minzi are also morphometrically very similar but H. minzi is bisexual. In addition, H. minzi was diagnosed with a more posterior position of the phasmids (2–4 annuli anterior to anus level). Comparing the morphometric data of paratypes to that of the published populations with linked DNA barcodes, a few differences are evident. For instance, the illustrations of Divsalar et al. (2020) show a relatively anterior position of the phasmids (7–11 annuli anterior to anus level) despite the 2–7 annuli number indicated in the authors’ descriptions. Furthermore, published data is based on a limited number of specimens (3 and 4), with one male recorded in one of the two populations. As already demonstrated by Subbotin et al. (2011, 2015), it is imperative that reference sequences from type material or from type locality-collected specimens be obtained as this will give a reliable sequence signature for these species and will provide a basis to clarify identification of the different existing population types (“Type A” and “Type B”), including resolving the taxonomic position of H. canadensis in relation to H. vulgaris.
In the current study, the ITS-rRNA gene evidently suffers from more substitution saturation and displays a much higher level of intraspecific polymorphism. However, the ITS region equally remains an excellent tool for diagnosing Helicotylenchus spp. as it showed concordant phylogenetic patterns to those of the 28S-rRNA gene. The COI gene sequences of the three species including the contentious H. microlobus are supplied herein for the first time. The taxonomic status of H. microlobus has already been discussed by Mwamula et al. (2020b) and Subbotin et al. (2015). By using a phylogenetic framework and morphological data of a H. pseudorobustus topotype population, Subbotin et al. (2015) demonstrated the distinctness and validity of the two species. Rybarczyk-Mydłowska et al. (2019) supplied COI barcodes of H. pseudorobustus populations whose 28S-rRNA barcodes are similar to that of the H. pseudorobustus topotype population. Similar to the 28S-, and ITS-rRNA gene analysis by Subbotin et al. (2015), the analysis of the COI gene sequences in the current study showed concordant phylogenetic patterns, with the two species standing out as genetically distinct, but morphologically cryptic species.
General remarks on molecular characterization
The genus Helicotylenchus comprises several valid but cryptic species. The diagnostic characters of these closely related species are marred by phenotypic plasticity with no well-defined boundaries (Subbotin et al., 2011, 2015). Linking molecular markers to morphological data is ideal as it augments and provides a better understanding of the morphologically unresolved species within the genus. However, in most cases, molecular taxonomists tend to extract DNA from random unmeasured specimens taken from larger target population(s) putatively identified based on morphometrics of a few specimens to represent the species of interest. But it is common for two or more Helicotylenchus species to co-exist as mixed populations. For instance, in the current study, H. caudatus and H. asiaticus populations (PJ2) were recovered from the same subsample in a ratio of 1:6. Random selection of specimens from such populations may result in amplifying DNA from unidentified species and erroneously linking such DNA barcodes to morphological data of rather a morphometrically identified species. We envisage this to be one of the main causes of incorrect linkages of morphospecies and molecular barcodes that frequently appear in the public databases.
Case studies of sequence mislabeling in other groups such as Pratylenchus spp. have already been detailed, including assigning sequence data of bacterial feeding Acrobeloides to a quarantine species Pratylenchus goodeyi (Janssen et al., 2017). Acrobeloides and Pratylenchus commonly occur as mixed populations in nematode communities (Lü et al., 2020). Unfortunately, apart from the obvious differences in stoma and pharyngeal corpus, some species of Acrobeloides such as A. nanus generally present body postures that are similar to Pratylenchus, including morphometric characters such as general body length, position of vulva, and tail shape, among others. It is therefore possible to mistake Acrobeloides individuals for Pratylenchus in a mixed population, especially when observed at a low magnification under reflected illumination of a stereo microscope that tends to obscure the details of the internal organs. We therefore suggest that morphometric data of specimens to be used in DNA barcoding should be taken prior to DNA extraction to avoid mis-linking barcodes of unidentified populations to putative species. In addition, as already noted by Janssen et al. (2017), several other factors are evidently implicated in contributing to the unsuitable nature of several sequences in public databases. For instance, several sequences contain PCR amplification or sequencing errors, cloning vectors, chimera sequences, unexcised primer sequences, and bioinformatics analysis errors, among others. Therefore, to ensure correct integrative nematode diagnosis, it is crucial to mitigate these potential pitfalls during the various steps involved in DNA barcoding to avoid the consequential cascade of erroneous interpretations in diagnosis.
Notes
Conflicts of Interest
No potential conflict of interest relevant to this article was reported.
Acknowledgments
This work was supported by Korea Institute of Planning and Evaluation for Technology in Food, Agriculture and Forestry (IPET) through Agriculture, Food and Rural Affairs Convergence Technologies Program for Educating Creative Global Leader Program, funded by Ministry of Agriculture, Food and Rural Affairs (MAFRA) (no.321001-03).