Introduction
Grapevine is affected by 63 viruses among which Grapevine fanleaf virus (GFLV) is one of the most destructive and worldwide distributed (Martelli, 2012). GFLV belongs to the genus Nepovirus in the family Secoviridae (Sanfaçon et al., 2009). It causes crop losses of up to 80% in various temperate regions (Andret-Link et al., 2004; Sokhandan Bashir et al., 2011). GFLV particles are polyhedral in shape with a diameter of 28 nm (King et al., 2011). The virus is transmitted in nature by the dagger nematode, Xiphinema index from one grapevine to another (Vigne et al., 2005), but also transmissible to experimental hosts such as Chenopodium amaranticolor and C. quinoa.
The genome of GFLV consists of two single-stranded positive-sense RNAs. Each RNA codes for a polyprotein denoted as P1 and P2 which are respectively expressed from RNA1 (Ritzenthaler et al., 1991) and RNA2 (Serghini et al., 1990). Protein 2B is one the three proteins encoded by RNA2 and acts as a movement protein (MP) which is a constituent of tubular structures occurring in modified plasmodesmata of GFLV-infected cells (Ritzenthaler et al., 1995; Serghini et al., 1990).
In the course of virus infection, there is a particular interest as to the understanding of whether or not the infection is localized and non-systemic. Most of the studies on virus MPs are focused on their function or loss of function through mutations, production of transgenic MP-expressing resistant plants, cellular localization, increase in the size exclusion limit of plasmodesmata, and interactions between MP and other cell proteins (Akamatsu et al., 2007; Deom et al., 1990; Haupt et al., 2005; Wolf et al., 1989). Also, MPs as non-structural proteins have contributed to antibody production and in vivo virus detection (Calegario et al., 2012; Cerovska et al., 2012; Salimi et al., 2010).
During the past decades, several serological methods were widely used to detect plant viruses together with molecular methods (Agrios, 2004; Hull, 2013). Unfortunately, traditional methods to raise antibodies against purified virus preparations have some limitations such as the low viral titer in the plant tissues and instability of the particles (Colariccio et al., 2000). Moreover, with such antibodies, serological cross reactivity occurs between closely related viruses especially between viruses belonging to the same genus (Hull, 2013). The use of viral proteins, such as non-structural protein MP, expressed in Escherichia coli as immunogens overcomes such problems (Fajardo et al., 2007). Obviously, most serological methods are based on coat protein (CP) because CP is highly conserved and thus facilities the detection of plant viruses by serological methods (Cerovska et al., 2006). On the other hand, a close serological relationship between GFLV and Arabis mosaic virus (ArMV) results in some cross reactivity in serological tests. To overcome this problem, detection by the use of antibodies prepared against a non-structural protein such as MP can be useful. Such an alternative has been used in detection of Potato mop-top virus (PMTV) because detection solely based on presence of PMTV CP seems to be inefficient as distribution of PMTV RNAs varies in different parts of infected plants and the multipartite virus. PMTV is capable of establishing infection in absence of the CP-encoding RNA and the putative cysteine-rich protein (Cerovska et al., 2006; Savenkov et al., 2003).
The recombinant viral non-structural proteins which are expressed in bacterial cells have great potentials as source of antigens to produce specific antibodies. As one of the strategies to advance studies on in situ detection of GFLV proteins, we expressed the GFLV MP in E. coli, produced specific polyclonal antibodies, and detected the MP in infected plants by the use of enzyme-linked immunosorbent assay (ELISA) and Western blotting.
Materials and Methods
Source of the MP gene
The GFLV MP gene was amplified by the specific primers GMPF1 (5′-GCGGAT GGNCG NACTACYGG-3′) and GMPR1 (5′-TCTCAY RGTCGARCTCAAWCKVGG-3′), cloned into pTZ57RT (Sokhandan Bashir et al., 2009), released by BamHI and SacI restriction digestion (Fermentas, Lithuania), and cloned in the expression vector pET21a to obtain pET-21aGFLVMP. The length of MP gene of GFLV is around 1,044 bp that cloned into pET21a.
E. coli DH5α was transformed with pET21aGFLVMP by the heat-shock method (Chung et al., 1989), and the transformants were selected on ampicillin. pET-21GFLVMP was purified using AccuPrep® Nano-Plus Plasmid Mini kit (Bioneer, Alameda, CA, USA) and sequenced with T7 promoter and terminator primers. Then, pET21GFLVMP was transformed into E. coli strain BL21 (DE3) for expressing the MP.
Expression in E. coli.
As a start culture, E. coli strain BL21 (DE3) containing pET21GFLVMP was grown overnight in Luria-Bertani (LB) medium containing 100 μg/ml ampicillin. Next, overnight culture was diluted 50 times in 10 ml of LB medium and grown (37°C, 200 rpm) until the optical density (OD600) was 0.4 to 0.6. Then, 1.5 ml of cell suspension was taken as non-induced control before isopropyl-β-D-thiogalactopyranoside (IPTG) was added into the culture at 1, 1.5, or 2 mM to induce the T7 promoter. Both induced and non-induced cell suspensions were grown in incubator with the same conditions as mentioned earlier. Samples from the incubated culture were analyzed after 4, 6, and 16 h of the induction by taking 1.5 ml of culture from the flask at each time. All cell suspension samples were centrifuged at 10,000 rpm for 7 to 10 min in a Heraeus Megafuge 1.0R rotor 3041 (Heraeus, Hanau, Germany).
Verification of expressed GFLV MP
After centrifugation of the culture sample, the pellets were re-suspended in Lysis buffer and the supernatant containing soluble proteins was subjected to analysis in sodium dodecyl sulphate polyacrylamide gel electrophoresis (SDS-PAGE) after centrifugation (Heraeus Megafuge 1.0R rotor 3041) at 10,000 rpm for 7 min. The remaining cell debris from pellet of induced culture was used for assessment of insoluble expressed recombinant protein. The pellets were kept at −80°C overnight and re-suspended by 1 × phosphate buffered saline (PBS) buffer and analyzed by SDSPAGE.
Western blotting
The extracted crude protein sample was mixed with an equal volume of 2 × Laemmli buffer pH 6.8 before boiling for 5 min to denature the protein. Then, 15 to 20 μl of each protein sample was separated on a 12% polyacrylamide gel containing SDS for 4 h at 100 V (Laemmli, 1970). The gel was stained with Coomassie brilliant blue G-250 (Thermo Fisher Scientific, Bremen, Germany) for 2 h and de-stained three times and each time for 2 h with the de-staining buffer (H2O 80%, methanol 10%, glacial acetic acid 10%). The protein bands were transferred onto nitrocellulose membrane using a wet blotting system (Akhtarian, Tehran, Iran). The trans-blotted membrane was incubated in blocking buffer (PBS + 2% bovine serum albumin) overnight, probed with 1:2,000 dilution of anti-His antibody in PBS (8 mM Na2HPO4, 1.5 mM KH2PO4, 2.7 mM KCl, 137 mM NaCl, pH 7.3), followed by incubation first with 1:5,000 dilution of rabbit anti-mouse immunoglobulin G (IgG)-alkaline phosphatase conjugate (Bioreba, Basel, Switzerland) and then with 1:5,000 dilution of nitrobluetetrazolium and 5-bromo-4-chloro-3-indolylphosphate (BCIP/NBT) substrate (Agdia, Elkhart, IN, USA) until the protein bands reached the desired intensity. The membrane was washed in deionized water for several minutes to stop the reaction and the membrane was air-dried on a filter paper and photographed.
Purification of expressed GFLV MP
After the expression and localization of GFLV MP fusion protein were confirmed, a colony of transformed E. coli strain BL21 (DE3) was inoculated into the protein expression medium as described previously and the cells were harvested by centrifugation at 10,000 × g for 10 min. The protein was then purified using pre-charged His-Bind Column (Novagen, Madison, WI, USA) under denaturing condition or His-Bind Column Chromatography kit (Novagen) as per manufacturer’s instruction. The soluble extract was loaded directly onto pre-charged His-Bind Column for purification. Urea was removed by dialyzing against several changes of PBS buffer (pH 7.3) with constant stirring at 4°C. Concentrations of the purified proteins were determined by Bradford’s method (Bradford, 1976) and Nano drop (Thermo Fisher Scientific).
Confirmation of purified recombinant GFLV MP
A 1 ml aliquot of the purified protein was re-suspended in 100 μl of Laemmli buffer, boiled for 5 min and 20 μl aliquots were loaded onto 12% polyacrylamide gel containing SDS (Laemmli, 1970). The protein purified from the columns was subjected to plate-trapped antigen ELISA (PTA-ELISA), as described previously (Dijkstra and de Jager, 1998) using anti-His-Tag as the first antibody and a universal horseradish peroxidase (HRP)-conjugated antibody as the secondary antibody. Finally, 3,3′-diaminobenzidine (DAB) was added as substrate of HRP to visualize the reactions.
Immunization of rabbits, absorption and calibration of antibodies
The purified recombinant MP was used as the antigen to raise antibodies in the two approximately 1.5-month-old New Zealand rabbits, four times with two weeks interval. The first injection was done subcutaneously with complete Freund’s adjuvant (1:1 v/v), and the three remaining injections with incomplete Freund’s adjuvant (1:1 v/v). Two weeks after the last injection, bleedings were carried out (40-50 ml/bleeding/animal). Blood samples were allowed to coagulate for 1 h at 37°C and 30 min at 4°C, and then centrifuged at 3,000g for 10 min. The supernatant (antisera) was aliquoted into microtubes and stored at −20°C.
Titration of the antiserum was done by the use of PTA-ELISA against the purified MP to determine its strength of reactivity and the optimal concentration. To achieve this, the antiserum was titrated with different dilutions in the range of 1:512 to 1:131,072. The immunoglobulin fraction was obtained from the antisera using Protein-A IgG Purification Kit (Thermo Scientific, Waltham, MA, USA). Antibody concentration was estimated on the basis of a specific extinction coefficient at A280 nm wavelength. IgGs were conjugated with alkaline phosphatase using Easy Link Alkaline Phosphatase Conjugation Kit (Abcam, Cambridge, UK) after neutralization of the purified IgG by a gel filtration-based desalting column.
The polyclonal antibodies developed against recombinant GFLV MP were evaluated for sensitivity and specificity by Western blotting, double antibody sandwich (DAS)-ELISA and PTA-ELISA. Crude leaf extracts from healthy and 12 samples of GFLV-infected tissues of grapevine and also purified MP from E. coli were used as test materials. Furthermore, a leaf sample of grapevine infected by ArMV used to checking the cross reactivity. Infected leaves by GFLV were collected from different regions of Iran include East-Azerbaijan, Zanjan, and Shahriyar. The RT-PCR by specific primers corresponding the CP region of GFLV confirmed that the leaves infected by GFLV.
Efficiency of the anti-GFLV MP antibodies was examined by PTA-ELISA following Dijkstra and de Jager (1998) and DAS-ELISA (Clark and Adams, 1977) to detect the purified MP and 12 samples of GFLV-infected plant tissues.
Western blot analysis
Infected and non-infected leaf tissues were ground in 1 × PBS, mixed with an equal volume of 2 × sample buffer and boiled for 5 min. Next, the denatured crude leaf extracts, purified GFLV MP and the protein from non-induced bacteria were separated by 12% SDS-PAGE, blotted onto nitrocellulose membranes using electro-blotter at 80 V for 120 min at room temperature as mentioned previously. After blocking with 5% powdered skim milk in phosphate buffered saline Tween 20 (PBST), the membrane was incubated with the anti-GFLV MP IgG (1:500) overnight at 4°C with gentle shaking. After 4 × washing with PBST, the membrane was incubated with the secondary antibody, mouse anti-rabbit IgG HRP-conjugate (1:1,000; Sigma-Aldrich, St. Louis, MO, USA) at room temperature for about 2 h. The bands of interest were visualized by reaction with freshly prepared DAB substrate (Roche Applied Science, Indianapolis, IN, USA).
This study was approved by the animal ethics committee of Pasteur Institute of Iran (IPI) (approval No. 66465132).
Results
Cloning and expression of GFLV MP
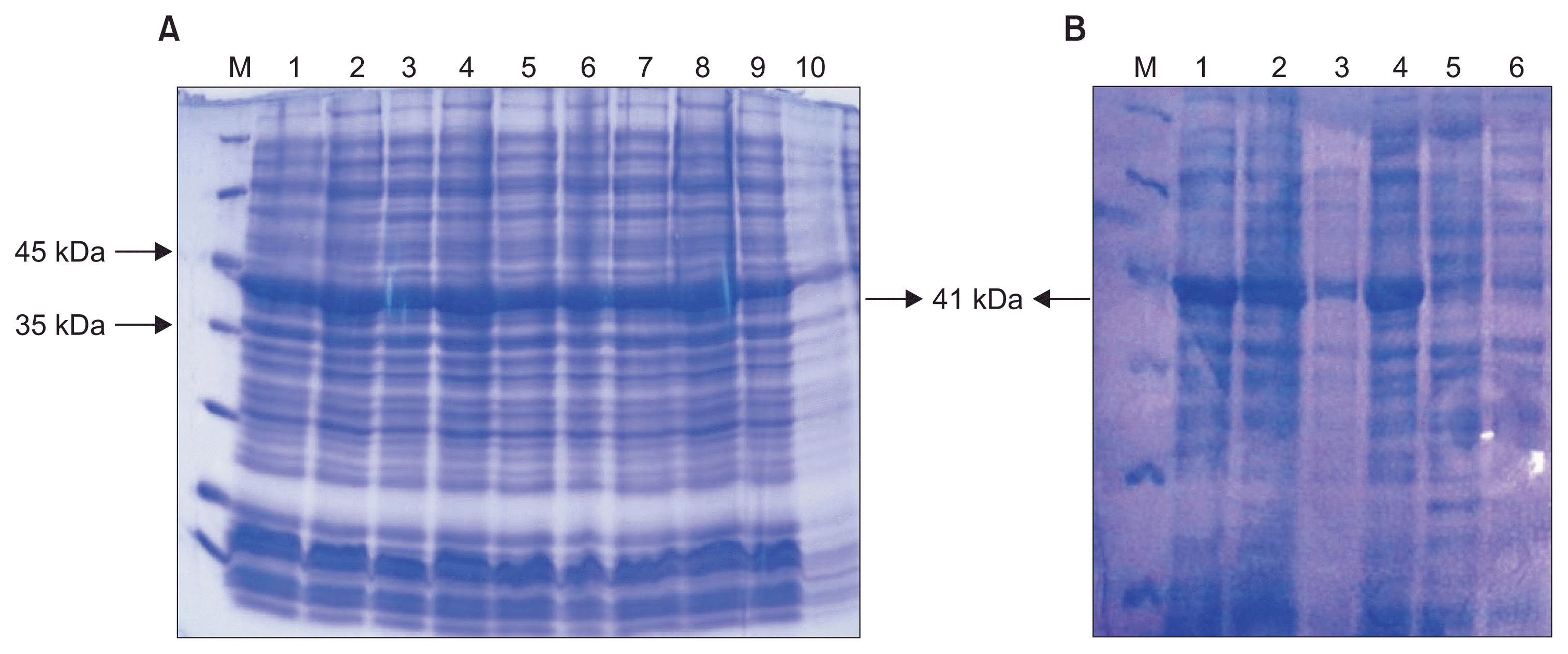
After ligation of the GFLV MP gene into pET21a and cloning into E. coli, the nucleotide sequencing data confirmed correct in-phase insertion of the full-length MP gene in the expression vector and possession of 6 × His-Tag at the C-terminal. The expression construct transformed into E. coli strain BL21 (DE3) for expressing the MP gene under different conditions which showed that there was no difference with regards to the level of the expression with respect to various concentrations of IPTG (1, 1.5, and 2 mM) and duration of induction (4, 6, and 16 h) (Fig. 1). Therefore, the expressing cells were harvested at 4 h after induction with 1 mM IPTG. Expression of the MP gene in E. coli was proved on 12% SDS-PAGE by an intense band with a size of about 41 kDa, which was roughly corresponded to the predicted size of the complete GFLV MP plus the fused amino acid tags. The anticipated band was mostly seen in the soluble fraction although a small amount of the protein was also visible in the insoluble fraction (pellet) (Fig. 1). Besides, extraction of the MP under denaturing condition at 6 M urea was higher than under non-denaturing condition (Fig. 1).
Fusion of a His-Tag at the C-terminal of the expressed protein resulted in an intense band at the expected position in the Western blotting with anti-His-Tag monoclonal antibody. Thus, it was concluded that it revealed band corresponded to MP (Fig. 2).
Purification and characterization of expressed GFLV MP
SDS-PAGE analysis of dialyzed purified protein showed an expected band of about 41 kDa corresponding to GFLV MP including the fused tags (Fig. 3). Concentration of the purified MP was estimated to be about 400 μg/ml, well above the optimal concentration of antigen for immunization of rabbit. Furthermore, the result of PTA-ELISA showed that the HRP-conjugated anti-His-Tag reacted with the in vitro expressed MP, demonstrating identity of the recombinant protein as the purified MP.
Efficiency of the antiserum and antibodies
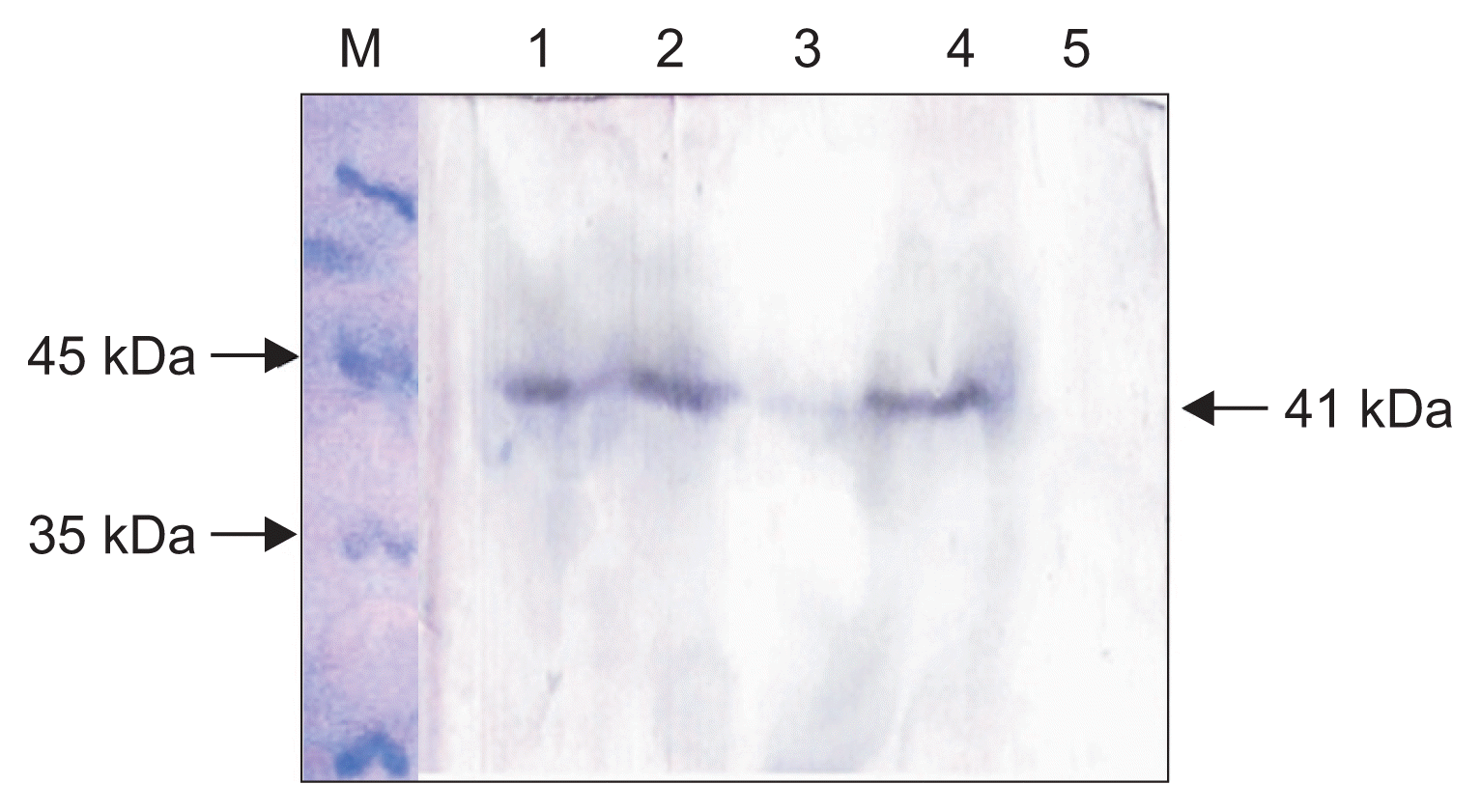
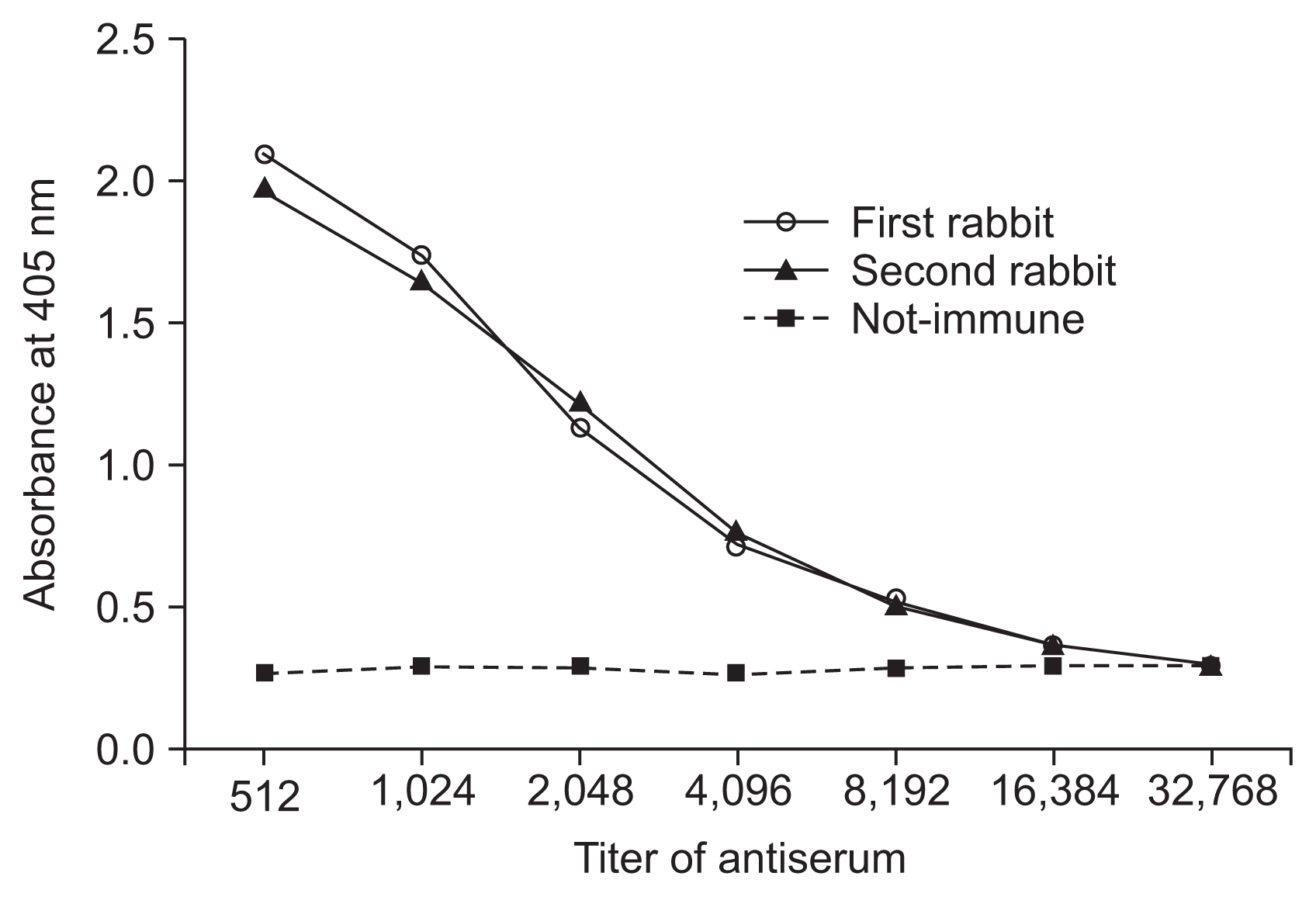
Results from titration by the use of PTA-ELISA revealed that the titer of the anti-GFLVMP antiserum was about 1:8,192 (Fig. 4). Extracts of grapevine leaves and purified MP diluted 1:10 and 1:5, respectively, were also recognized using 1:2,000 dilution of anti-MP serum (Fig. 5). However, no significant reaction was observed in extracts from negative control samples such as healthy plant and the protein extracted from non-induced bacteria.
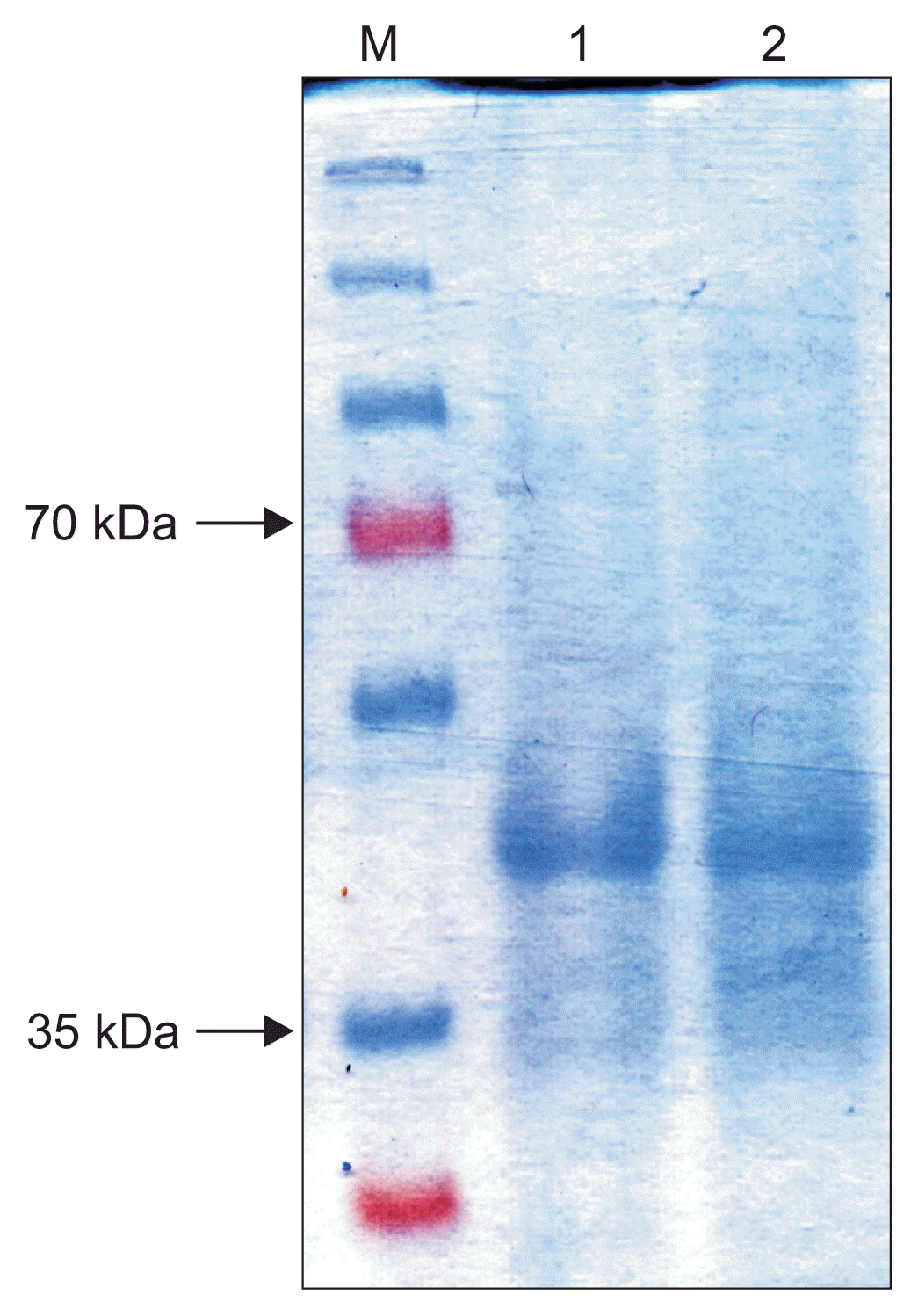
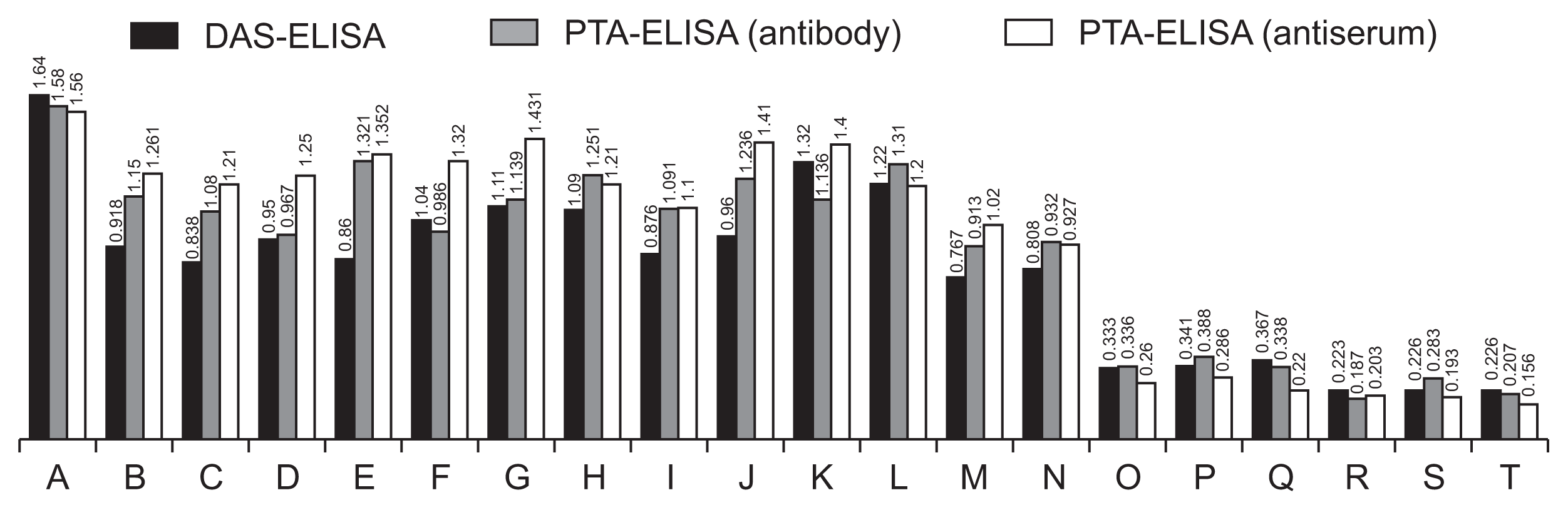
The purified anti-GFLV MP IgG at 1:500 dilution detected GFLV in the infected grapevine leaves (Supplementary Table 1) and reacted with the purified expressed MP (Fig. 5). Results from DAS-ELISA which was applied to evaluate efficiency of the prepared anti-GFLV MP IgG and the conjugated IgG (both at 1:500 dilution) to detect the antigens showed that both IgG and the conjugate reacted efficiently with the expressed and purified protein as well as with GFLV in the infected leaf tissue. However, the absorbance at 405 nm with the infected plant material was lower than that with the expressed protein (Fig. 5). Moreover, the antibody reacted with the expressed protein as well as the extract from GFLV-infected grapevine leaves at 1:500 antibody dilution in Western blotting, but the signal was weak, probably due to low concentration of MP in the plant tissue. However, no signal was observed with the crude extracts from the healthy plants (Fig. 6).
Discussion
Diagnosis and control of plant viruses constitute an imperative and relevant necessity (Lima et al., 2012). Over the last few decades, laboratory-based methods have been developed and used routinely in many laboratories for accurate diagnosis of diseases caused by plant viruses. These techniques involve physical, biological, cytological, serological, and molecular properties of viruses. Although various laboratory methods have been developed and adopted for virus diagnosis serological methods are still one of the most specific and robust methods to achieve a rapid and precise virus detection (Astier et al., 2001; Lima et al., 2012; Naidu and Hughes, 2003).
The purpose of the current research was to examine the possibility of using in vitro expressed GFLV MP, instead of GFLV CP or the virions, to prepare antigenic protein and its application in producing polyclonal antibodies. Similarly, other investigations have successfully used recombinant expressed virus proteins to prepare antibodies for the detection of a number of plant viruses (Abou-Jawdah et al., 2004; Cerovska et al., 2003, 2006, 2012; Hourani and Abou-Jawdah, 2003; Iracheta-Cárdenas et al., 2008; Jain et al., 2005; Jelkmann and Keim-Konrad, 1997; Korimbocus et al., 2002; Kumari et al., 2001; Lee and Chang, 2008).
Expression of the complete GFLV MP gene as a fusion protein with His-Tag was successfully achieved by induction with 1 mM IPTG for the duration of 4 h. It is noteworthy that most of the expressed proteins are in the soluble fraction which is similar to what have been reported by others (Hema et al., 2003; Hourani and Abou-Jawdah, 2003; Lee and Chang, 2008; Raikhy et al., 2007). Nevertheless, a little amount of MP was also detectable in the pellet fraction in our study.
The expressed protein was analyzed by SDS-PAGE and the presence of a band with a molecular mass of approximately 41 kDa, the expected size for the GFLV MP, was observed. About 3 kDa difference with the in vitro MP mass was due to fusion of the six histidine tag to the recombinant MP. Alternatively, identity of the expressed protein was confirmed by Western blotting (Fig. 1). It is worthy to be that no significant effects were reported in the immunogenicity of recombinant antigens as compared to the conventional antigens (Kumari et al., 2001; Mutasa-Gottgena et al., 2000).
Recombinant viral non-structural proteins expressed in bacterial cells have great potential as a source of antigens for raising specific antibodies. In the case of PMTV, antibody based on the virus MP has been obtained for diagnostic purposes because the CP is not required for infection by this virus; therefore, detection of the virus with the CP-based antibody would not be efficient (Savenkov et al., 2003). Thus, the detection based on non-structural proteins could be useful when combined with other detection methods. As such, the use of recombinant proteins as the immunogenic material is an attractive strategy for the preparation of antibodies against viruses which occur in low concentrations in infected plants, or difficult to purify. On the other hand, the use of antibodies against the recombinant structural proteins in diagnostic tests seems to be impeded by their inefficiency in recognizing native epitopes. Alternative detection techniques such as tissue blot immunoassay or dot immunobinding assay-ELISA may be needed to overcome such a difficulty (Korimbocus et al., 2002). Moreover, anti-MP antibodies can be used in studies on functionality of this protein in infected plant and its role in the viral movement within plant tissue.
As mentioned earlier, different dilutions of the polyclonal antibodies were tested to find out an optimal concentration for the detection with anti-GFLV MP. The results indicated that the MP can be detected in GFLV-infected plant and in purified preparation from E. coli by PTA-ELISA. The optimal conditions for the diagnosis included 1:10 dilution of the leaf extract, 1:1,000 dilution of the anti-MP serum and 1:500 dilution of anti-GFLV MP IgG.
Additionally, anti-GFLV MP IgG and the conjugate efficiently detected the MP and GFLV in the infected plant tissue when DAS-ELISA was applied (both diluted at 1:500). Although there are reports on antibodies prepared against recombinant viral proteins which do not detect the related virus in DAS-ELISA (Cerovska et al., 2006; Korimbocus et al., 2002) it was not the case in this study. Such inefficiency could be due to inability of the coated antibodies to react with native viral epitopes.
The purified expressed MP and GFLV-infected plant extract were both efficiently detected by the anti-GFLV MP IgG in Western blotting assays although a non-specific background reaction was also observed (Fig. 6). Notwithstanding, such non-specific reactions are not unusual in the blottings where polyclonal antibodies to recombinant proteins are used (Cerovska et al., 2003; Gulati-Sakhuja et al., 2009; Hourani and Abou-Jawdah, 2003; Kumari et al., 2001). There are suggestions to address the non-specific reactions that point to premature or partial transcription of target gene, and high hydrophobicity of the MP (Cerovska et al., 2003; Chen et al., 2000). The MP is associated with the host cell membranes and binds the high histidin repeats to antibody (Gulati-Sakhuja et al., 2009; Hourani and Abou-Jawdah, 2003).
In this study, antibody prepared against GFLV MP was demonstrated to be efficient in the detection of either the purified MP or the virus in ELISA and Western blotting. Availability of such antibodies facilitates screening of plant material in sanitation schemes and being implicated in studies on pathogenicity of the virus.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print