Introduction
The members of the Xanthomonas genus are Gram-negative and motile with a single flagellum, and produce a yellow pigment called xanthan. Xanthomonas species cause serious and devastating diseases in over 400 plants, including economically and agriculturally important crop species such as tomato, pepper, soybean, and rice (Ryan et al., 2011). Among them, Xanthomonas campestris pv. vesicatoria (Xcv) is the causal agent of bacterial spot disease on tomato and pepper, and the disease causes serious reductions in yield and quality (Bouzar et al., 1994). This pathogen has been used extensively as a model system to elucidate molecular mechanisms of virulence in plant-microbe interactions (B├╝ttner et al., 2003). In addition to Xcv, X. axonopodis pv. glycines (Xag) causes bacterial pustule disease, which is one of the most serious diseases of soybean worldwide. Symptoms are typically characterized by small, yellowish-brown areas with a raised pustule. Xag can cause a severe loss of yield in conditions favorable to the pathogen (Hong et al., 2012).
Methylation of DNA is a process where methyl groups (CH3) are transferred to DNA by DNA methyltransferases, resulting in the production of modified DNA. DNA methyltransferases are present in all domains of life (Razin and Riggs, 1980). In eukaryotic organisms including humans and plants, methylation of cytosine producing 5-methylcytosine (5mC) is mainly found, and is involved in the epigenetic regulation of gene expression (Laird, 2010). In addition to 5mC, N6-methyladenine (6mA) as well as N4-methylcytosine (4mC) are predominant modifications in bacterial genomes (S├Īnchez-Romero et al., 2015). The 5mC, 6mA, and 4mC modifications are precisely mediated by DNA methyltransferases recognizing specific motifs in bacterial genomes. These DNA base modifications are known to be involved in a wide range of bacterial processes, such as expression and regulation of genes, regulation of the cell cycle, phase variation, maintenance of genes, and resistance to bacteriophage infection (Haagmans and van der Woude, 2000; Kahramanoglou et al., 2012; Labrie et al., 2010; Low et al., 2001; Skarstad and Katayama, 2013).
To understand the functions of DNA methylation, methylation patterns and motifs in an entire genome (the methylome) have to be determined. The 5mC base modification has been detected by bisulfite DNA sequencing in eukaryotic organisms (Frommer et al., 1992). However, this method is limiting for studying bacterial DNA methylation because it is difficult to detect 6mA and 4mC, which are abundant in bacterial genomes (Casades├║s and Low, 2006). A state-of-the-art technique, single-molecule real-time (SMRT) sequencing, has been developed to determine genome sequences as well as to detect modified nucleotides including methylated DNA (Flusberg et al., 2010). SMRT sequencing can identify methylated DNA modifications including 6mA and 4mC in real-time using fluorescent labeled nucleotides and single polymerase molecules (Eid et al., 2009). Using this technique, diverse bacterial methylomes including those of Helicobacter pylori, Lactobacillus spp., Mycobacterium tuberculosis, Escherichia coli, and Campylobacter coli have been determined, and previously unreported DNA methylation motifs have been identified (Lee et al., 2015; Powers et al., 2013; Zautner et al., 2015; Zhang et al., 2015; Zhu et al., 2016). Plant diseases caused by Xanthomonas spp. are agriculturally and economically important, and the genus has been extensively studied at the molecular and genomic levels as a model system for host-microbe interactions. However, methylome analysis of Xanthomonas spp. has not been reported.
Here, the methylomes of two important Xanthomonas spp., Xag strain 8ra and Xcv strain 85-10, whose genome sequences have been previously reported (accession Nos. AM039952 and JDSU00000000, respectively) (Lee et al., 2014; Thieme et al., 2005), were characterized by SMRT sequencing techniques. We identified 6mA and 4mC modification in the two genomes, and assigned DNA methylation motifs including previously unreported methylation motifs. Comparison of putative DNA methylation motifs from the two species revealed that each strain has a unique DNA methylation system.
Materials and Methods
Bacterial growth and genomic DNA preparation
Xcv strain 85-10 and Xag strain 8ra were grown and maintained at 28┬░C in tryptic soy medium (tryptic soy broth, soybean-casein digested: 30 g/l). For genomic DNA extraction, Xanthomonas was cultured in XVM2 medium (Wengelnik and Bonas, 1996). The medium contained 20 mM NaCl, 10 mM (NH4)2SO4, 5 mM MgSO4, 1 mM CaCl2, 0.16 mM KH2PO4, 0.32 mM K2HPO4, 0.01 mM FeSO4, 10 mM fructose, 10 mM sucrose, and 0.03% casamino acids (pH 6.7). Bacterial cells grown to OD600 = 0.6 were collected by centrifugation, and washed twice with 1 ml of 50 mM Tris-HCl (pH 7.8). Genomic DNA for SMRT sequencing was then extracted with the DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany).
Single-molecule real-time sequencing
Libraries for SMRT sequencing were prepared using 5 ╬╝g genomic DNA from each strain. The SMRTbell template libraries were constructed with SMRTbellŌäó Template Prep Kit 1.0 (100-259-100; Pacific Biosciences, Menlo Park, CA, USA) following the manufacturerŌĆÖs instructions. For 20 kb libraries, we additionally used the BluePippin DNA Size Selection System to remove smaller fragments of SMRTbell template (< 20 kb). Sequencing primers were annealed to the SMRTbell templates followed by binding with the complex using the DNA/Polymerase Binding kit P6 with the MagBead loading kit (Pacific Biosciences). The SMRTbell library was sequenced using one SMRT cell (Pacific Biosciences) with C4 chemistry (DNA sequencing Reagent 4.0), and 240 min movies were taken for each SMRT cell using the PacBio RS II instrument (Varela-├ülvarez et al., 2006).
Bioinformatic analysis of SMRT sequencing data
SMRT sequencing reads of the two different Xanthomonas strains were assembled using the Hierarchical Genome Assembly Process (HGAP, version 2.3) workflow developed at Pacific Biosciences (Chin et al., 2013). During this process, assembled contigs were polished with PacBio subreads using Quiver to remove sequencing errors. Finally, contig accuracy was checked by MUM-mer 3.5 (Kurtz et al., 2004) for identifying the bacterial genome and plasmids, with trimming of one of the two similar ends for genome closure. Assembled contigs were structurally and functionally annotated using Prokka, a stand-alone tool specifically developed for bacterial genome annotation (Seemann, 2014).
Genome-wide base modification and analysis were performed using the default settings in the SMRT Analysis 1.1 and the RS_Modification_and_Motif_Analysis 1 protocol (Pacific Biosciences). Briefly, fluorescent signals were quantitated kinetically as part of the SMRT sequencing process. DNA modifications altered the kinetic characteristics of time for base incorporation and measured changes in interpulse duration (IPD). The IPD ratio was calculated by comparing the observed IPD to the in silico IPD control, and a base modification quality value (QV) score was calculated as the Phred transformed P-value of detection at each position in the genome. QV score threshold was set at 30 for genome-wide methylation pattern analysis. Motif identification was performed using MotifMaker (https://github.com/PacificBiosciences/MotifMaker) and in-house Python scripts with REBASE version 608 (ftp://ftp.neb.com/pub/rebase/allenz.txt). Identifying where the methylation motifs were located, we developed in-house Python scripts which concatenate open reading frames (ORFs) for predicting hypothetical operon and predicted promoter regions using PePPER (de Jong et al., 2012). Subsequently, Python scripts found the position of the methylation motifs, and calculated the methylation ratio for the promoter, gene body and intergenic regions.
Nucleotide sequence accession number
All sequence data have been deposited in the National Center for Biotechnology Information (NCBI) database. Accession numbers of Xag strain 8ra are CP017188 and CP017189 for the chromosome and for the plasmid pXAG_1, respectively, and accession numbers of Xcv strain 85-10 are CP017190, CP017191, CP017192, and CP017193 for the chromosome and for the plasmids pXCV_1, pXCV_2, and pXCV_3, respectively (Table 1).
Results and Discussion
Through SMRT sequencing, DNA modifications of the Xag strain 8ra genome were analyzed and the positions of modified DNA bases (4mC and 6mA) were determined. Totals of 66,682 and 4,147 modified bases showing a QV score of over 30 were detected as 4mC and 6mA, respectively, in Xag (Table 1). Interestingly, most methylated bases were found in the chromosome rather than in plasmids. Among the total numbers of 6mA and 4mC methylated bases, 6mA methylated bases were not detected and two of the 4mC methylated bases were observed in the native plasmid (Table 1). In support of this observation, the native plasmids of Xag did not contain any putative DNA methyltransferase (Kim et al., 2006). This may be one reason that very few methylated bases were detected in the plasmid. Alternatively, it is possible that Xag strain 8ra has recently adopted the native plasmid.
Next, we identified putative methylation motifs by using the REBASE database (Roberts et al., 2010) and MotifMaker (PacBio). The modified bases with flanking regions (20 bp length) to either side were used for identifying putative motifs. Fifty-three putative motifs showing at least 50 methylated bases for each motif were predicted in the Xag genome (Supplementary Table 1). The ten most prevalent methylated motifs are shown in Table 2. Among the top 10 motifs, three motifs were known as sequences recognized by specific restriction enzymes (NotI, FnuDII, and MwoI) in other species, but seven motifs were putative new methylation motifs, indicating Xag may possess novel methyltransferases compared with previously known DNA methylation systems. Although the numbers of 4mC methylated bases were significantly higher (16-fold) than that of 6mA, six and four motifs corresponded to 4mC and 6mA, respectively, in the top 10 motifs. Among these motifs, two motifs showed that over 50% of the sites were methylated, and both were 6mA methylation. CAGNNNNNNNNTCTY was the most highly methylated motif (80.42%) under the growth conditions used here.
DNA modifications of the Xcv strain 85-10 genome were also characterized by the same methods used for Xag. In the case of Xcv, 2,321 4mC methylated bases and 5,491 6mA methylated bases were identified with a QV score of over 30 (Table 1). In contrast to Xag, where the majority of methylated DNA modifications were identified as 4mC, in Xcv the number of 6mA methylated bases was over 2-fold higher than that of 4mC methylated bases. Interestingly, the total number of methylated bases detected in Xcv was considerably lower than in Xag. Although both strains belong to the Xanthomonas genus, it is clear that their methylation patterns are dramatically different and it may be that their methylation systems have recently diverged or evolved after speciation. Similar to methylation patterns in Xag, most methylated DNA bases were found in the chromosome of Xcv rather than in the plasmid. Among 2,321 4mC methylated bases, 2,233 and 88 were located in the chromosome and native plasmids, respectively. In the case of 6mA, 5,135 were detected in the chromosome and 356 in native plasmids (Table 1). This means that the native plasmids of Xcv possess significantly higher numbers of methylated bases compared to those of Xag, which had only two detected methylated DNA bases in the plasmid. Xcv native plasmids are reported to contain three putative DNA methyltransferases (Thieme et al., 2005). These are likely responsible for the different methylation patterns in native plasmids between the two bacteria.
To identify putative methylation motifs in Xcv, the methods used for Xag were employed. Xcv possessed 50 possible methylation motifs, each showing over 50 detected methylation modifications (Supplementary Table 2). Among the top 10 methylation motifs in Xcv (Table 3), three were likely new motifs. Interestingly, except for two motifs (CCCGGG and CCNNGG), eight were identified as 6mA. Among the five known motifs (Table 3), CCCGGG is recognized by SmaI restriction enzyme. Yu and Yang (2007) reported that Xcv strain 7-1 has M.XveII, which is a DNA methyltransferase that specifically methylates the second cytosine residue in CCCGGG. Xcv strain 85-10 also contains a homolog of M.XveII, XCV1110 (Thieme et al., 2005). Because XCV1110 is identical with M.XveII at the amino acid level, methylation of CCCGGG in Xcv strain 85-10 is likely to be catalyzed by XCV1110. Unlike Xag, six putative methylation motifs in Xcv were highly methylated (over 80%) under the given conditions. Except for these six motifs, all other putative motifs described in Table 2 and Supplementary Table 2 showed below 4% of methylated bases in each motif.
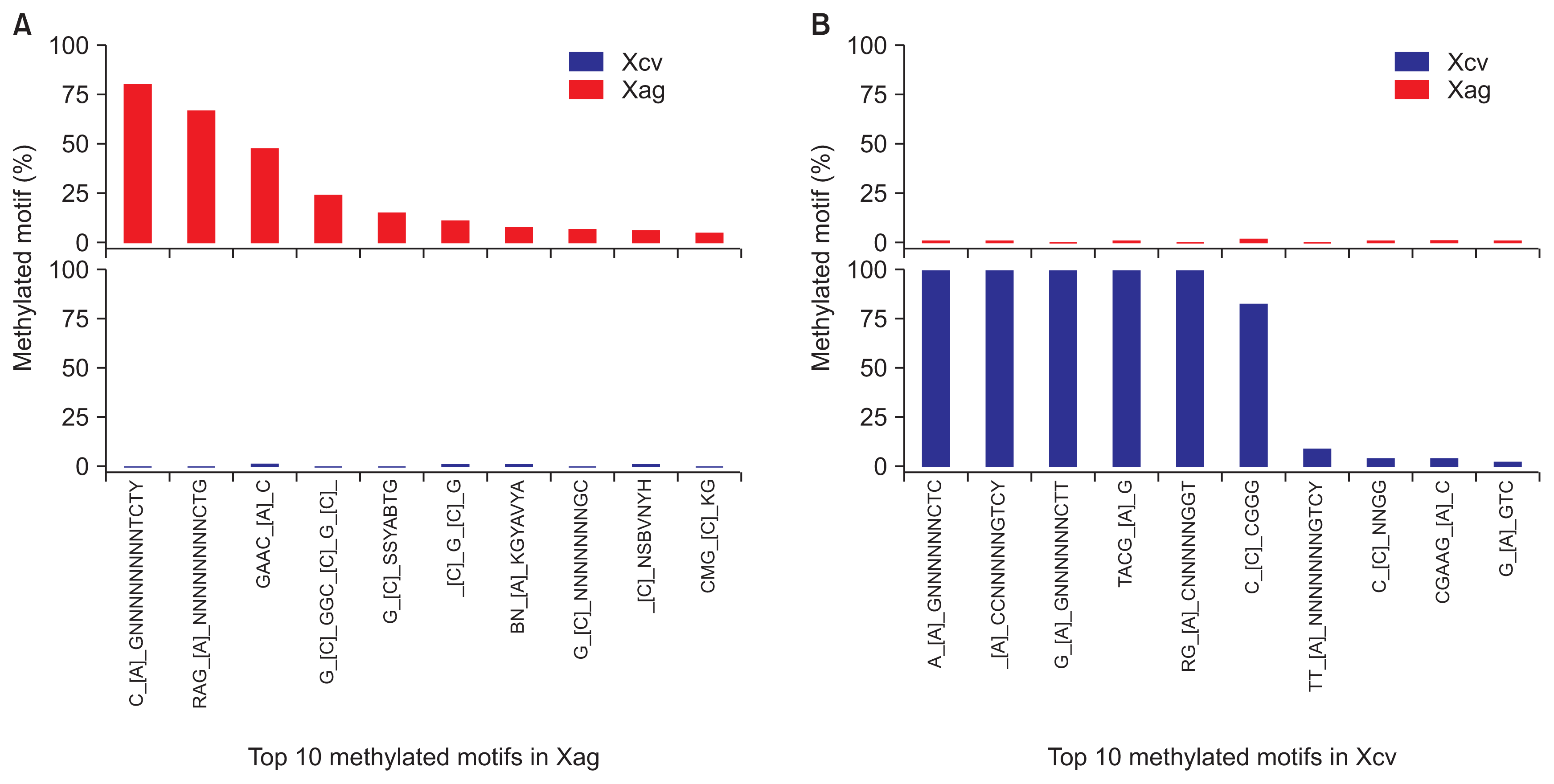
Next, we compared the top 10 methylation motifs from Xag and Xcv. Although Xag and Xcv belong to the genus Xanthomonas, methylation motifs of Xag were dramatically different from those of Xcv. There were no common or shared motifs in the top 10 methylation motifs of each strain (Table 2, 3). The top 10 methylation motifs in Xag were detected in the Xcv genome, but the percentages of motifs bearing methylated residues in Xcv were at almost undetectable levels compared to those in Xag (Fig. 1A). Likewise, the top 10 methylation motifs of Xcv were also found in Xag, but the proportion of their methylation in Xag was considerably lower than in Xcv (Fig. 1B). Although the top 10 methylation motifs from each strain were detected in both Xag and Xcv, there was no meaningful correlation of methylation patterns and motifs between the two Xanthomonas spp. under the given conditions. These results suggest that they possess species-specific DNA methylation systems. Species-specific DNA methyltransferases in each bacterium may have been adopted after speciation of the genus Xanthomonas during evolution.
We further investigated the distribution of the top 10 methylation motifs at the genome level (Fig. 2). The genomes of both strains were divided into three regions: putative promoters predicted by the PePPER program, gene bodies (open reading frames and operon regions), and intergenic regions, defined as any region in the genome excluding putative promoter and gene body regions. Among 10 motifs in Xag, six motifs were mainly distributed in gene bodies and intergenic regions (Fig. 2A). Four motifsŌĆöCAGNNNNNNNNTCTY, RAGANNNNNNNNCTG, GAACAC, and BNAKGYAVYAŌĆöshowed that at least 1% of methylated bases were located in putative promoter regions. Methylation of these four motifs may be involved in regulation of gene expression. The Dam site (GATC), which is widely distributed in the entire E. coli genome, plays a role in regulating phase variation. The methylation status of a Dam sites upstream from the pap operon is critical for the binding of Lrp regulator and expression of the pap operon (Nou et al., 1993). Likewise, over 1% of methylated bases of five motifs (AAGNNNNNNCTC, GAGNNNNNNCTT, TACGAG, RGACNNNNNGGT, and GAAGAC) in Xcv were detected in putative promoter regions (Fig. 2B). Interestingly, base modification of all nine promoter-associated motifs from both Xag and Xcv was 6mA but not 4mC. In addition to Dam methylation, CcrM (GANTC) methylation is known to be associated with regulation of gene expression in bacteria (Low et al., 2001). Both Dam and CcrM methylation are 6mA base modifications. Therefore, nine motifs that were detected in putative promoter regions and involved 6mA base modification are possibly related to gene regulation control in Xanthomonas spp. Although distribution of putative DNA methylation motifs was presented in this study, roles of methylation in gene body are difficult to speculate. Genome-wide distribution and patterns of methylated motifs have recently been determined by SMRT sequencing and roles of methylation in gene body are not well-documented at genome levels in other bacteria. Subsequent studies will be required for elucidating functions of individual DNA methyltransferases and their motifs at genome-wide levels in Xcv and Xag.
The bacterial strains used for methylome analysis in this study were cultured in XVM2 media where Xanthomonas spp. could express diverse virulence-related genes and secrete major virulence factors for infection (Wengelnik and Bonas, 1996). However, as mentioned above, the methylation patterns of Xag and Xcv were dramatically different. They may possess different virulence factors and their responses to identical conditions are likely different because their host ranges are clearly distinct and they have specialized life cycles. Xag infects soybean but cannot infect tomato and pepper, and Xcv causes severe disease in tomato and pepper but not soybean. These differences may have an influence on the distinct methylation patterns observed in identical media and incubation conditions, because regulation of gene expression in phase variation and virulence by DNA methylation is well established (Camacho and Casades├║s, 2002; Casades├║s and Low, 2006; Marinus and Casadesus, 2009; S├Īnchez-Romero et al., 2015). In addition, DNA methylation is one of the known antiviral mechanisms of bacteria (Labrie et al., 2010). Within the same bacterial species, different strains show distinct resistant patterns against infection of diverse bacteriophages (Chae et al., 2014). Host ranges of bacteriophages are most likely determined by specific DNA methyltransferases in bacteria. Therefore, it can be easily speculated that Xag and Xcv possess different resistant ranges against bacteriophage infection and the dramatically different methylation patterns in two Xanthomonas spp. may be one of ways to explain species- or strain-specific antiviral responses against phage infection. The results presented in here can provide knowledge relevant to bacterial resistance against bacteriophage infection.
In this study, we analyzed and compared the 6mA and 4mC methylomes of two Xanthomonas spp. using SMRT sequencing techniques. We demonstrated preliminary DNA methylation patterns that further showed possible species-specific methylation motifs. The epigenetic regulation of gene expression in Xanthomonas spp. is little studied. The employment of SMRT sequencing techniques has led to new insight into understanding the biology and functions of DNA methylation in this genus. Genome analysis of both Xag and Xcv strains revealed that they contain 15 and 10 putative DNA methyltransferases, respectively (Lee et al., 2014; Thieme et al., 2005), and there is no significant homology except XAR_0064 (Supplementary Table 3). Although we show putative methylation motifs of Xag and Xcv in this study, we still do not understand the functions of specific DNA methyltransferases including their recognition sites in both species. To figure out their functions and importance at genome levels, the roles of individual DNA methyltransferases are under investigation by combination of SMRT sequencing techniques and biochemical/phenotypical analysis.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print