Bacterial leaf blight caused by Xathomonas oryzae pv. oryzae, bacterial grain rot caused by Burkholderia glumae and Acidovorax avenae subsp. avenae are major bacterial diseases and also seed-borne pathogens of rice (Goto, 1964; Kadota, 1996; Shakya et al., 1985). Especially, the occurrence of bacterial leaf blight and bacterial grain rot is becoming more common in Korea as the weather is getting hot and humid (Jeong et al., 2003) and these pathogens can be a threat to rice production in both temperate and tropical rice growing regions due to its high epidemic potential (Ham et al., 2011). But most of all, seedborne bacterial pathogens are of particular concern because, unlike seedborne fungi, strategies for the management are inadequate, especially given the limited and antiquated chemical options available (Gitaitis and Walcott, 2007). Also, year-to-year transmission resulting from the seed-borne nature of the pathogen may also contribute to yield losses (Trung et al., 1993). Moreover, infected seeds may be discolored, but more often no conspicuous symptoms are visible (Song et al., 2004). Rice seeds infected with these pathogens are important sources of primary inoculum and a means of dissemination to new areas. The spread of seed borne pathogens has a negative effect on rice yield, which is why sensitive and simple detection methods are required to screen. Accurate diagnosis and identification of plant pathogens is a pre-requisite of disease management to sustain high yield potential of crops. Therefore continuous efforts are being made to develop a simple, reliable, rapid and safe method for the disease diagnosis (Sharma, 2003). Although methods have been developed to detect seed-borne bacterial pathogens, none have been optimized to selectively detect the bacterium from rice seeds. Recently, several studies have used polymerase chain reaction (PCR) technology as a tool to detect and study the variability of pathogenic bacteria (Xiang and De Boer, 1995). Among PCR assays, multiplex PCR has been developed to simultaneously detect several plant diseases (Asano et al., 2010; James et al., 2006), thus providing a reliable, fast, and inexpensive method for routine detection of plant bacterial diseases in the field. Therefore it is very important to develop mPCR assay to detect three bacterial pathogens in rice seeds.
Based on the 16S and 23S rDNA sequences for A. avenae subsp. avenae and B. glumae, and the transposase A gene sequence (AE013598.1, REGION:466077..47102) for X. oryzae pv. oryzae, specific primers were designed for in vitro DNA amplification (Table 1). Since three bacteria have to be detected at a same time, primers were designed to produce three different sizes of PCR products represent individual bacterial species and to use same annealing temperature for the optimum condition for PCR amplification. With the 63┬░C as an optimum annealing temperature, three sets of primers (Og-F/R, XOT-F/R, and Oal-F/R) had been designed to produce 402 bp for B. glumae, 490 bp for X. oryzae, and 290 bp for A. avenae subsp. avenae. In order to confirm the right sizes of PCR products, three isolated bacteria had been tested for the multiplex PCR amplification. Bacterial DNA was harvested from 1 ml cells (OD600, 0.8-1.0) and extracted using the Genomic DNA extraction kit (iNtRON, Seongnam, Korea) based on the protocol for Gram-negative bacteria. The concentration of extracted DNA was determined by measuring the absorbance ratio of 260:280 nm using a NanoDrop spectrophotometer (Nanodrop ND-1000; NanoDrop Technologies Inc., Wilmington, DE, USA). The multiplex PCR assay was performed using the Rice Bacteria PCR Detection Kit (IncloneTM, our latest patented kit; Inclonebiotech Co., Seoul, Korea) following the manufacturerŌĆÖs protocol with minor modifications. Briefly, the final volume of the multiplex PCR mix was adjusted to 20 ╬╝l with sterile distilled water, in which the reaction mixture contained three sets of specific primers, template DNA, reaction buffer, dNTP mixture, and enzyme mix. The cycling parameters included an initial denaturation step at 95┬░C for 15 min followed by 35 cycles of 95┬░C for 20 s, 63┬░C for 30 s, and 72┬░C for 30 s, ending with a final extension at 72┬░C for 5 min. Amplified products were detected on a 1.5% Tris-borate-ethylene-diaminetetraacetic acid (EDTA) agarose gel with loading star (Dyne Bio, Seongnam, Korea). Multiplex PCR using six primers suggested in this study amplified the expected targets for B. glumae, X. oryzae pv. oryzae, and A. avenae subsp. avenae, respectively (Fig. 1). Each amplification product at a specific size was observed for the three bacteria: B. glumae (402 bp), X. oryzae pv. oryzae (490 bp), and A. avenae subsp. avenae (290 bp). Nevertheless single or triple, right sizes of bands had been produced without any trivial bands. Therefore, these primer sets can be used for the multiplex PCR to detect three major bacterial pathogens, B. glumae, X. oryzae pv. oryzae, and A. avenae subsp. avenae, in rice at a same time.
In order to test the specificity of these primer sets, PCR amplification had been performed with 20 different bacterial DNA including three different pathovars of X. axonopodis, four different X. campestris pathovars, X. oryzae pv. oryzicola, Pseudomonas fluorescens, P. aeruginoa, two P. savastanoi pathovars, P. cochorii, P. viridiflava, five P. syringae pathovars and Rhizobium sp. (Table 2). None of those bacterial DNA produced right products by the mPCR amplification using primer sets in Table 1. However, all tested five different strains of B. glumae or four different races of X. oryzae pv. oryzae produced the expected bands for both bacterial species (data not shown). Therefore, primer set Og-F and Og-R is a specific for B. glumae and primer set Oal-R and Oal-R is a specific for A. avenae subsp. avenae. In the case of primer set XOT-F and XOT-R was able to produce right products for X. oryzae pv. oryzae but not for X. oryzae pv. oryzicola, this primer set may be a pathovar specific.
To avoid the isolation of DNA from bacteria and determine the sensitivity of PCR detection in pure culture suspensions, direct multiplex PCR had been conducted (Noh et al., 2012). Bacterial suspensions were prepared as serial 10-fold dilutions to 10ŌłÆ8. Briefly, each bacterial suspension of 106 to 107 cfu/ml in 1 ml sterile distilled water was diluted eight times in a 10-fold series. Aliquots (3 ╬╝l) of the dilutions were directly used for PCR amplification. According to the manufactureŌĆÖs protocol, direct multiplex PCR was performed in a 20 ╬╝l volume, in which the reaction mixture contained three sets of specific primers, three types bacterial suspensions, buffer, dNTPs, and enzyme mix. Detection limits of direct multiplex PCR from bacterial suspensions were tested using 10-fold (10ŌłÆ1 to 10ŌłÆ8) serial dilutions. Positive results were obtained using direct multiplex PCR at dilutions from 10ŌłÆ2 to 10ŌłÆ4 with primers for B. glumae, X. oryzae pv. oryzae and A. avenae subsp. avenae (Fig. 2). In contrast, faint PCR products were obtained at 10ŌłÆ5 to 10ŌłÆ8 dilutions, while no amplification was obtained in the uniplex (Fig. 2B, C) or multiplex assays beyond a 10ŌłÆ5 dilution (Fig. 2B). The 10ŌłÆ2 to 10ŌłÆ4 dilutions were the most effective concentrations for simultaneous direct detection of three bacteria (Fig. 2A, B). These results indicate that the direct multiplex PCR method developed in this study could be used to rapidly detect pathogen however, serial dilutions are essential to evaluate the sensitivities of direct multiplex PCR assays.
The primer sets designed to detect simultaneously three bacterial pathogens, B. glumae, X. oryzae pv. oryzae and A. avenae subsp. avenae, showed specificity and sensitivity enough to be used for multiplex PCR assay. However, all tests done above had been conducted with pure cultured bacteria. Though the PCR methods can be very useful to detect bacterial pathogens in pure culture, sometimes there are limitations to detect bacteria directly from seeds or plants because of the presence of some inhibitory compounds. To confirm that test these methods can be applicable to detect bacterial pathogens on seeds, multiplex PCR assay had been done using rice seeds infected artificially with three bacterial combinations. Surface sterilized rice seeds with 70% ethanol were soaked in bacterial culture with 108 cfu/ml concentrations for 48 h. The hulls were removed from the soaked rice seeds and direct multiplex PCR assay had been done with infected hull rice (Fig. 3). Single amplicons were detected in samples infected with a single bacterium: B. glumae (lane 1), X. oryzae pv. oryzae (lane 2), and A. avenae subsp. avenae (lane 3) and multiple amplicons were detected in seeds subjected to double and triple infections: B. glumae + X. oryzae pv. oryzae (lane 4), B. glumae + A. avenae subsp. avenae (lane 5), X. oryzae pv. oryzae + A. avenae subsp. avenae (lane 6), and X. oryzae pv. oryzae + B. glumae + A. avenae subsp. avenae (lane 7).
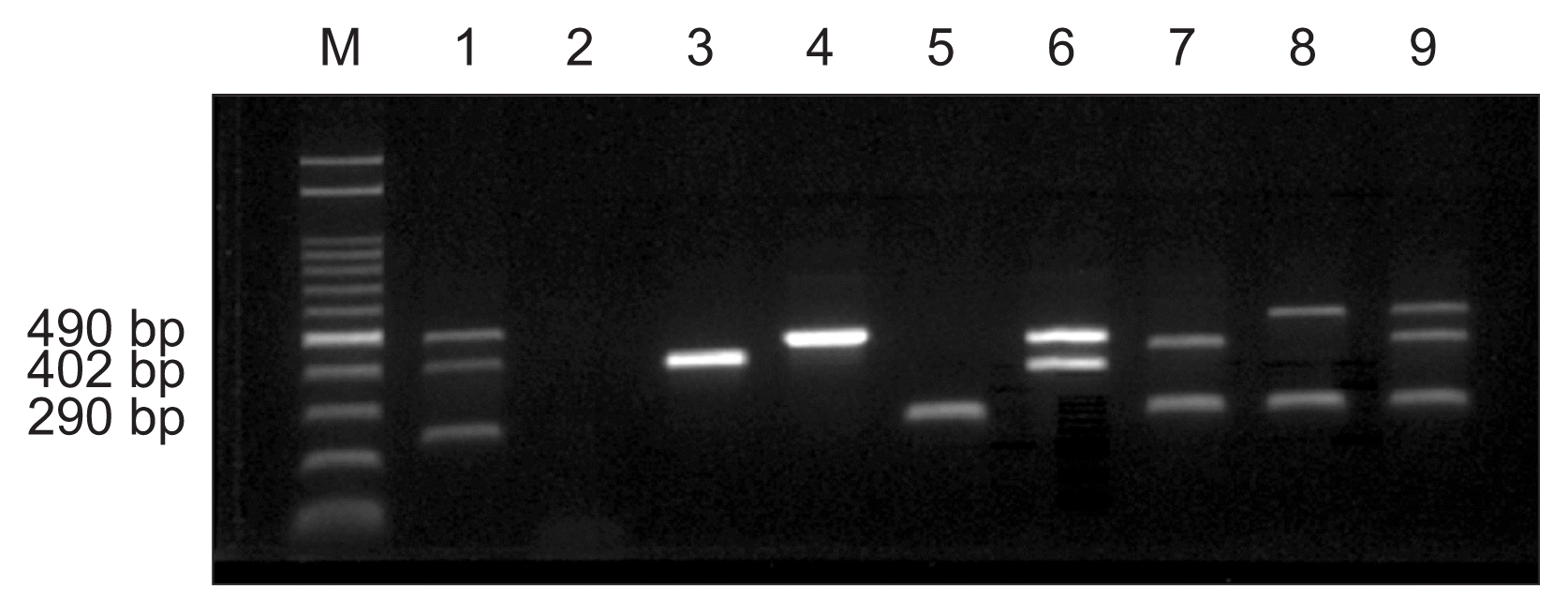
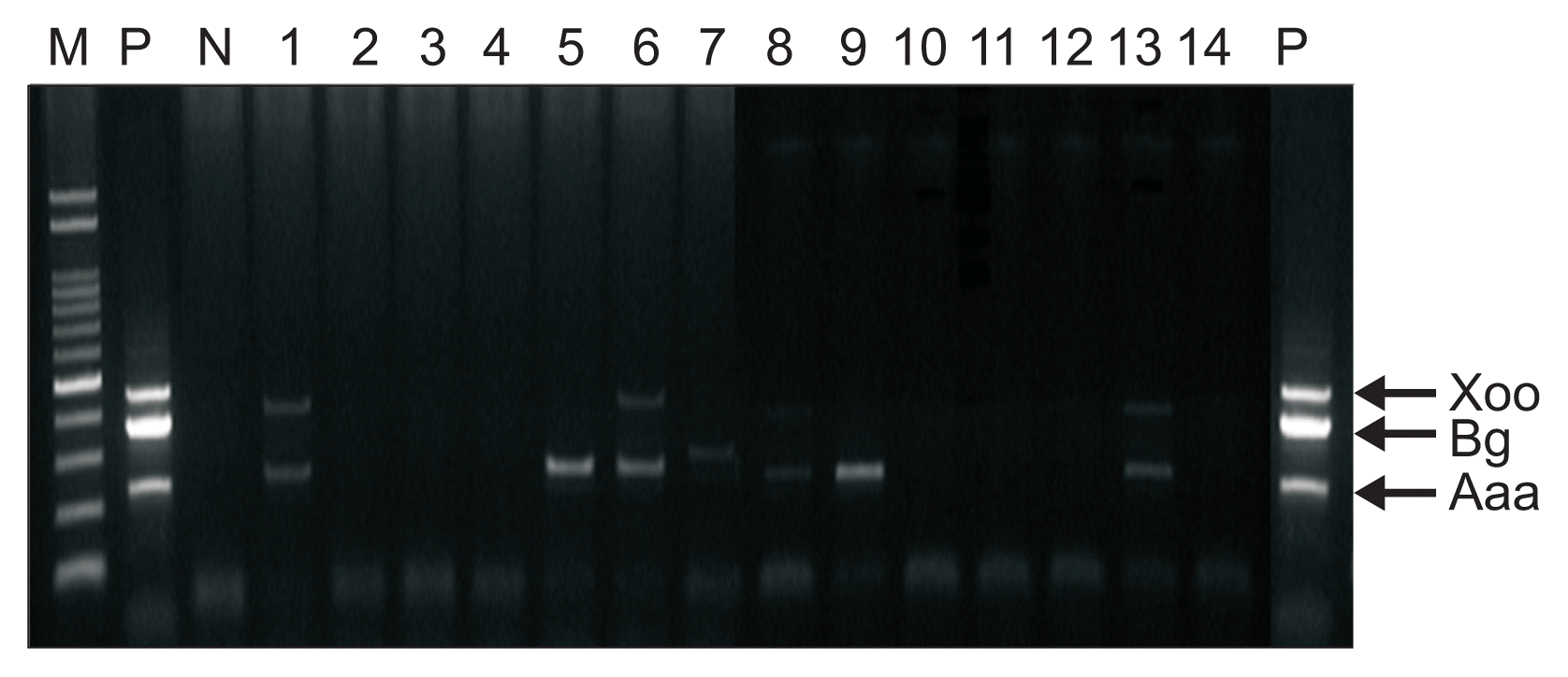
Since direct mPCR assay with artificially infected rice seeds had been done successfully, naturally infected rice seeds were tested. To detect bacterial pathogens in naturally infected rice seeds, head of rice plants showing symptoms collected from several provinces in Korea and hull rice of each samples were examined using the direct multiplex PCR method (Fig. 4, 5). Four out of total 14 samples were infected with two bacteria, B. glumae and A. avenae subsp. avenae (Fig. 5; lanes 1, 6, 8, and 13) and three samples were infected with A. avenae subsp. avenae only (Fig. 5; lanes 5, 7, and 9). X. oryzae pv. oryzae was not detected at all. These data were confirmed with bacteria isolated from symptomatic plants (data no shown).
Overall, the method can provide a specific and convenient tool for detecting B. glumae, X. oryzae pv. oryzae and A. avenae subsp. avenae in infected rice seeds.



 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print