Colletotrichum acutatum and Glomerella cingulata (anamorph C. gloeosporioides) both cause anthracnose diseases of strawberry (Fragaria ├Ś ananassa), resulting in major economic damages to strawberry production worldwide (Curry et al., 2002; MacKenzie et al., 2006; Smith and Black, 1990; Ure├▒a-Padilla et al., 2002). C. acutatum is known to cause mainly fruit-rot and spots on leaf, petiole and runner of strawberry while G. cingulata is associated with crown-rot leading to plant death. However, the fungicides available for use are not very effective because the disease develops rapidly after latent infection stage, and only a few resistance genes in strawberry have been identified (Denoyes-Rathan et al., 2005; Lerceteau-K├Čhler et al., 2005). In Japan, strawberry anthracnose by G. cingulata is incidental while C. acutatum is frequently detected (Kikuchi et al., 2010). Because strawberry seedlings are vegetatively propagated from the runners of mother plants, keeping the new clonal plants free from anthracnose is one of the most important steps to prevent the diseases. For this purpose, the nursery plants should be geographically isolated from the farmerŌĆÖs fields where anthracnose is often found. The northern Hokkaido island in Japan is one of such places for the seedling production because the epidemic of G. cingulata has not been reported yet. However, because latent infection of G. cingulata in mother plants brought from other areas was often found (Sumino et al., 2009), it is important to detect and eliminate mother plants infected by G. cingulata. Although the symptoms are quite distinct when appeared on plants (Fig. 1), infected plants normally have a latent period of infection from fungus invasion to first sporulation, which is an important factor for the disease spread in a strawberry field because symptomless foliages become a significant source of inoculum for subsequent crown and fruit infection (Leandro et al., 2001; Legard, 2000). It is therefore critical in preventing anthracnose diseases of strawberry whether we can detect the fungi from asymptomatic, healthy-looking plant parts when the invading fungus is in a quiescent stage.
For diagnosis of anthracnose, because the morphological characters of fungus and plant symptoms vary and thus cannot be used as a reliable tool, molecular techniques such as enzyme-linked immunosorbent assay (ELISA) and polymerase chain reaction (PCR) have been used to identify Colletotrichum species. PCR-based methods have been successfully used for detecting many fungal pathogens including Colletotrichum species in total DNA extracted directly from symptomatic plant tissues (e.g., lesions) (Freeman and Katan, 1997; Freeman et al., 2001; Garrido et al., 2009; Mart├Łnez-Culebras et al., 2000; Parikka and Lemmetty, 2004; Sreenivasaprasad et al., 1996). Using artificially and naturally infected plants, Parikka and Lemmetty (2004) previously showed that a PCR-based method could be used to detect C. acutatum if good-quality DNA was prepared from plant tissues. They also demonstrated that their PCR method could be used to detect C. acutatum in plant parts that did not show visible symptoms. In fact, there are not many reports that describe detection of the fungi from healthy-looking plant parts in latent infection (Parikka and Lemmetty, 2004) although it is very important to protect strawberry from anthracnose before the disease spreads in nursery plants. However, considering that the fungus may be localized in particular tissues in an initial stage of infection, one question is raised: Can we really judge that the plant is not infected when we do not detect the fungus from all tested plant parts? Even though we sample the plant part containing the fungal spores or hyphae, the fungus quantity must be very low and may be below the detection limit. In addition, with a simple PCR method, we may detect even dead spores that do not contribute to anthracnose. The other problems in application of a simple PCR method to anthracnose diagnosis are difficulties in removing PCR-disturbing inhibitors from strawberry tissues and judging faint or non-specific PCR products. Because PCR-disturbing inhibitors such as polyphenol compounds are very abundant in DNA extracted from strawberry tissues, unless we use a commercial DNA extraction kit specially designed for plants like strawberry, the quality of DNA is not necessarily high enough for stable PCR reactions. In a simple PCR method, we often experienced some problems that a detected band with the expected size was very faint or a few extra bands affected the results. Although we could establish the best DNA isolation and PCR method, and design a perfect primer pair for PCR in laboratories, we often encountered these problems especially when we have to handle thousands of plants for diagnosis. To solve these problems of PCR, we here developed the PCR method complimented with microtube hybridization (MTH); the method is designated PCR-MTH.
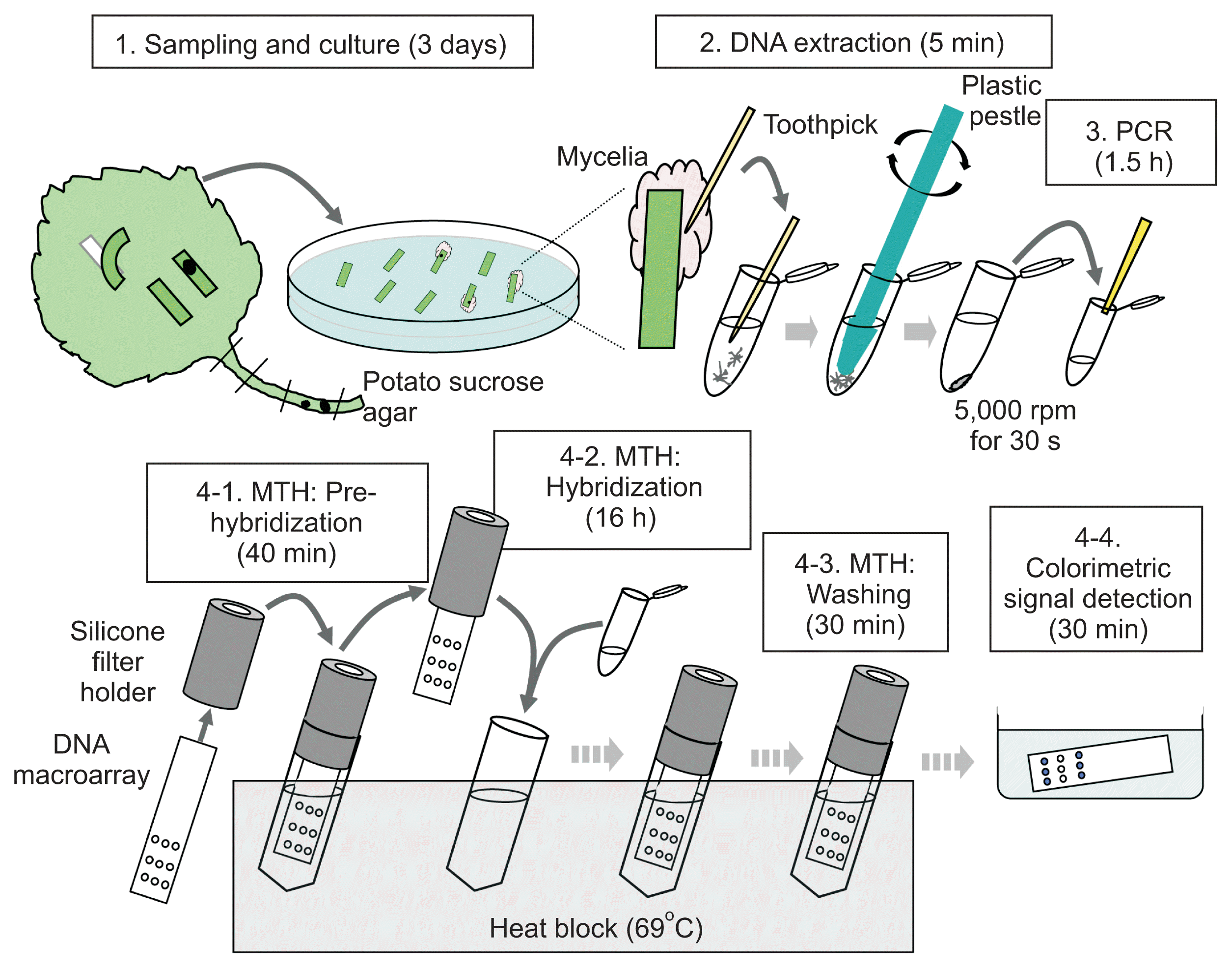
All the plants including mother plants to produce a nursery used in this study were prepared by Hokusan Co., Ltd. (Ishikari, Hokkaido, Japan) and grown in the greenhouse and fields of the same company. The sequential procedure of the diagnostic method developed in this study is outlined in Fig. 2. For the sample preparation, an outer petiole at a lower position was sampled from each plant because such petioles were located near the soil surface and likely to be exposed to soil-borne pathogens. The sampled tissues were then washed with running water. After washing, the tissues were cut out in the appropriate size (e.g., 10 ├Ś 3 mm), and each piece were surface-sterilized with 3% sodium hypochlorite solution for 10 min. The sterilized pieces were put on the potato sucrose agar including 0.01% streptomycin sulfate salt and incubated at 25┬░C for 3 days. Three days after inoculation (Step 1), hyphae were scraped off with a sterilized toothpick, put into 100 ╬╝l of water or TE buffer (10 mM Tris-HCl, 1 mM ethylenediaminetetraacetic acid, pH 8.0) in 1.5-ml microtubes and homogenized with a disposable plastic pestle (Funakoshi, Tokyo, Japan), manually (Step 2). After centrifugation at 5,000 rpm for 30 s, the supernatant was directly used for the following PCR as a fungal DNA template. One microliter of fungus DNA solution was added to 19 ╬╝l of the PCR reaction mixture (Step 3). The reaction mixture contained 2.5 units Tth DNA polymerase (Roche Diagnostics, Tokyo, Japan), 1├Ś PCR buffer, 1 mM dNTP (Roche Diagnostics) and 0.5 ╬╝M of each primer. The forward primer was biotin-labeled at the 5ŌĆ▓ end for colorimetric signal detection. The PCR conditions consisted of 1 cycle of 2 min at 94┬░C, followed by 35 cycles at 94┬░C for 30 s, 56┬░C for 30 s, and 72┬░C for 30 s. For specific identification of C. acutatum and G. cingulata by multiplex PCR, the primers were designed for specific regions in the gene MAT1-2, which codes for a factor determining mating type. A primer pair to amplify a small subunit rDNA region that is highly conserved among fungi was also added to the multiplex PCR primer set as a positive control (White et al., 1990). The primer sequences and sizes of amplified fragments are summarized in Table 1.
For the preparation of DNA macroarray, each fragment PCR-amplified with the specific primers was cloned and sequenced. Then, the pathogen-specific sequence was re-amplified with the same primers, and the amplified products (5 ng) were spotted in triplicate on a strip of nylon membrane (40 ├Ś 10 mm), Biodyne plus (Pall, Tokyo, Japan), by high-throughput liquid handling systems with glass syringes (Shieh et al., 2002). To prepare such a macroarray, we can either ask a supplier (e.g., Hokusan, Hokkaido, Japan) to make a macroarray or prepare a handmade macroarray by ourselves; there are commercially available kits to make a hand-made array such as DNA Microarray Making Kit (Funakoshi, Tokyo, Japan). To denature the DNA on the solid phase, we incubated the membranes at 120┬░C for 30 min in a dry incubator. The probes were immobilized by irradiation (120 mJ/cm2) at 254 mm in a ultraviolet cross-linker (UVP, Upland, CA, USA). We here describe briefly the general steps in MTH although they can be easily modified as the users like. A strip of nylon membrane with DNA is inserted into the slit of a silicone folder, put into the 2-ml microtube containing 1.7 ml of preheated PerfectHyb hybridization solution (Toyobo, Tokyo, Japan) and incubated at 69┬░C for 40 min (pre-hybridization). Transferring a membrane with a silicon holder from one tube to another is actually the trick in MTH, which makes the hybridization step very laborsaving; how to make the silicon holder has been described in Shimura et al. (2015). During this incubation, the PCR products from each sample are denatured at 99┬░C for 5 min and immediately placed on ice. After pre-hybridization (Step 4-1), the membrane held by the silicone filter folder is transferred into the new tube containing 1.7 ml of the preheated PerfectHyb hybridization solution (Toyobo) plus 15 ╬╝l of the denatured PCR products, and incubated at 69┬░C overnight (hybridization). After hybridization (Step 4-2), the membrane with a silicon holder is transferred to the new tube containing the primary wash buffer (2├Ś saline sodium citrate [SSC], 0.1% sodium dodecyl sulfate [SDS]) and incubated at 69┬░C for 1 min. This primary washing (Step 4-3) is repeated twice more at different time periods (1 min and 10 min). Then, the membrane with a silicon holder is transferred to the new tube containing the secondary wash buffer (0.1├Ś SSC, 0.1% SDS) and incubated at 69┬░C for 1 min. This secondary washing is also repeated twice more for different periods (1 min and 10 min). After the washing step, several membranes are rinsed together in 15 ml of the blocking solution (5% SDS, 125 mM NaCl, 25 mM sodium phosphate, pH 7.2) in a 50-ml centrifugation tube at room temperature (RT). The membranes are then incubated in 15 ml of the fresh blocking solution for 5 min, transferred to 15 ml of fresh blocking solution containing 10 ╬╝l of streptavidin alkaline phosphatase conjugate (New England Biolabs, Tokyo, Japan) and incubated for 5 min at RT. After two rinses in solution I (1:10 dilution of blocking solution), the membranes is rinsed twice in solution II (10 mM NaCl, 1 mM MgCl2, 10 mM Tris-HCl, pH 9.5) for 5 min at RT. Finally, the membranes is immersed in 10 ml of the solution for colorimetric signal detection (0.4 mg/ml nitro blue tetrazolium, 0.19 mg/ml bromo-4-chloro-3-indoryl phosphate, 50 mM MgSO4, 100 mM Tris-HCl, pH 9.5) in a plastic container (Step 4-4). After 1 min, the membranes are wrapped with plastic wrap and incubated for 15 min to visualize the positive spots at RT.
In this diagnosis, we added a step of culturing fungi to avoid the risk that we fail to collect the plant tissues containing the living fungus in latent infection. In addition, it is better not to use any organic solvents and expensive DNA extraction kits for the DNA extraction from strawberry tissues. A piece of tissue sample sterilized by a sodium hypochlorite solution is placed on a culture plate and incubated at 25┬░C for 3 days. If we observe fungal hyphae on the tissue and later identify as Colletotrichum species, then we judge that the original plant had been latently infected.
The gene MAT1-2 that codes for a factor determining mating type was chosen to design the specific PCR primers for G. cingulata and C. acutatum identification. Because the sequence of MAT1-2 gene is highly differentiated among species and well conserved within species, we were able to design very specific primers for each fungus based on the results of our sequencing and those of Du et al. (2005). However, because we did not want to rely on a simple PCR with electrophoresis, which depends on the sizes of PCR products for judgment, we replaced electrophoresis with a hybridization step to obtain the sequence information of the target. In MTH, all steps of pre-hybridization, hybridization and washing can be simply done in 2-ml microtubes on a heat block at 69┬░C.
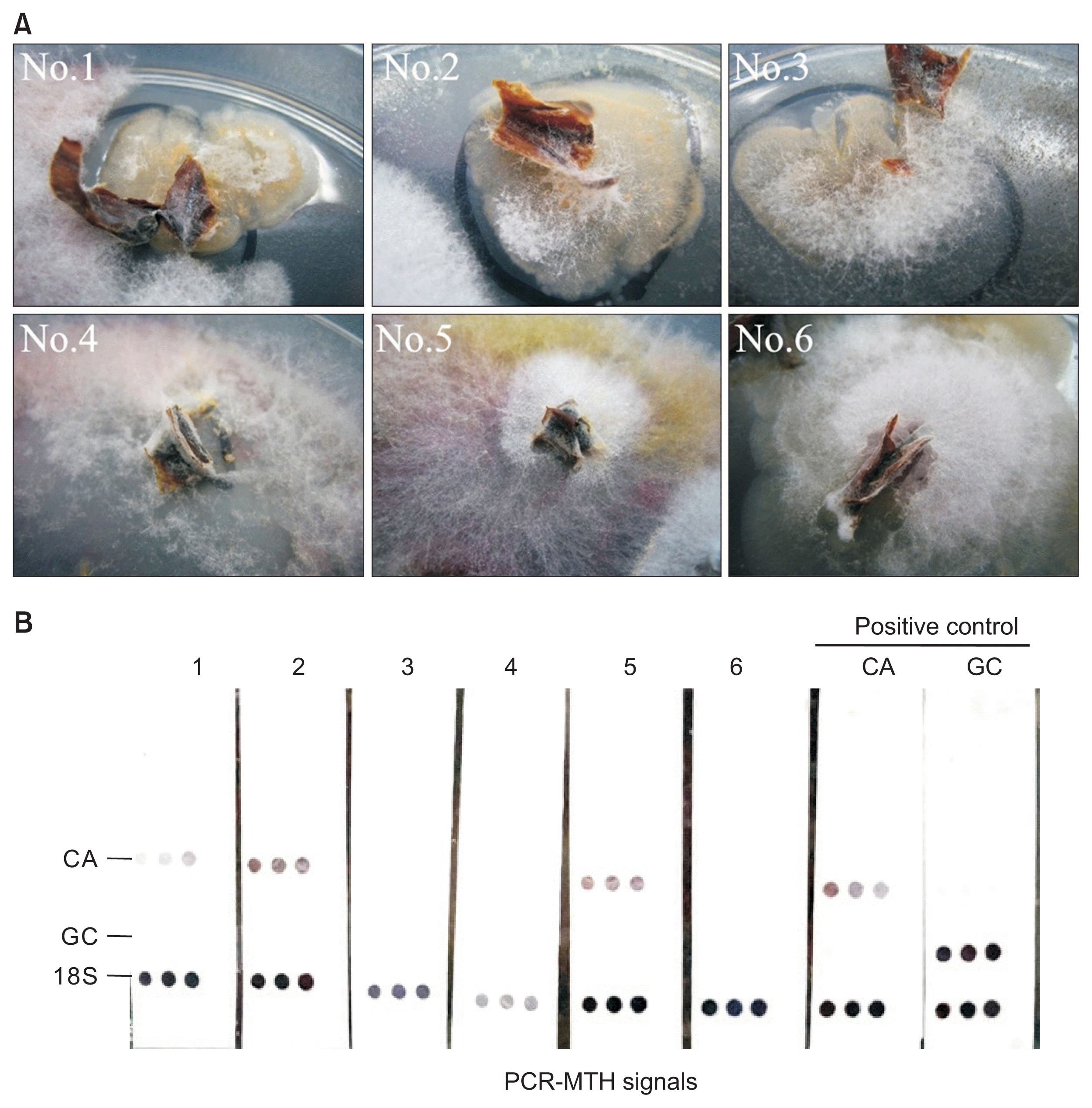
In order to evaluate if the methods developed here is really useful for detection of latent infection of anthracnose especially for practical application, we conducted a large scale survey using a few thousands of healthy-looking nursery plants of strawberry for three consecutive years from 2013 to 2015 (Table 2). Fig. 3A showed some examples of the fungal colonies obtained after initial culturing. Although nos. 1, 2, and 5 were identified as C. acutatum by PCR-MTH, their morphologies were similar to those of other fungal samples; it was indeed difficult to identify the fungi based on their morphology. In 2012, we conducted a preliminary survey to determine the experimental conditions by trial and error for the diagnosis using PCR-MTH. Out of 1,825 healthy-looking strawberry nursery plants, we could identify 4 C. acutatum-positive plants by PCR-MTH although we may have failed to catch all the infected plants due to our immature skills. Most fungal colonies, which showed negative signals, were later found to be Verticillium spp. In 2013, along with the system we fixed in 2012, we embarked on the project to completely eliminate latent infection of anthracnose fungi from our nursery fields. A piece of petiole was sampled from every 2,048 healthy-looking plants and placed on a culture plate after surface sterilization. Fungi were grown from 30-40% of those samples. Among them, 171 colonies were first selected as possible anthracnose fungi based on the color and feature of myceria; 21 out of the 171 colonies were eventually judged as a positive by PCR-MTH (Table 2). In the second year 2014, 218 colonies were selected in the same way; however, all of them were found to be as a negative by PCR-MTH (Table 2). Thus, we could finally eliminate latent infection of anthracnose from the nursery fields in Hokusan Co. Also in the third year, we were not able to find any latent infection. This success suggests that continuous monitoring of the fungi is very important to eliminate anthracnose. In this project, our farmers learned that they should watch plants regularly and remove potentially-infected plants as much as possible. We feel that the PCR-MTH method manifests its power especially for large-sale screening for latent infection of anthracnose. Because in Hokkaido, Japan, we are on alert for epidemics of G. cingulata, which causes a more severe anthracnose than C. acutatum, a reliable and simple diagnostic method for latent infection is really needed.
Several PCR-based methods have been developed to detect the anthracnose disease in Japan (Kikuchi et al., 2010; Sumino et al., 2009). Each of these methods has demerits as well as merits. For example, one method needs an expensive DNA extraction kit, another uses an additional nested PCR step, and others cannot distinguish G. cingulata and C. acutatum. Although all the conditions for PCRs including design for specific primers are well set up at the experimental (laboratory) stage, they do not necessarily work well as the number of tested plants becomes high. Perhaps the quality of extracted DNA from strawberry may not be good enough when the assay is scaled up. In addition, electrophoresis-based judgment cannot provide any sequence information. A simple PCR with electrophoresis does not seem to be perfect, considering that thousands of nursery plants must be tested in a relatively small laboratory without decent equipment in the frontline of strawberry production. We thus reasoned that the PCR products should be confirmed by their nucleotide sequences, perhaps by hybridization, which had been originally developed for viral detection in mixed infection (Sugiyama et al., 2008).
Toward application to a practical field, the PCR-MTH has been successfully introduced in the strawberry nurseries in the Ishikari and Abashiri regions of the Hokkaido island in Japan since 2011, where the farmers by themselves monitor the latent infection of anthracnose fungi by the PCR-MTH method. Because the epidemic of G. cingulata has not been reported yet in Hokkaido, it is important to prepare themselves for invasion of the fungus. When some G. cingulate-positive signals were detected in Abashiri, Hokkaido by the PCR-MTH method, those suspected plants were immediately removed just in case.
Verticillium wilt caused by a soil-borne pathogen Verticilium dahliae is also one of the most important diseases in strawberry production. Because it is important to use pathogen-free soil and a fast, sensitive detection of the pathogen from plants and soil for effective prevention, some PCR-based techniques have been developed. For example, Kuchta et al. (2008) demonstrated that a ribosomal RNA gene could be used as a specific marker to diagnose V. dahliae. To further develop our PCR-MTH method, which was tested for anthracnose here, as a versatile diagnostic tool for strawberry diseases, the application to Verticillium will be certainly the next target.
In conclusion, we proved that our system for nursery production using the diagnostic method for latent infection of anthracnose could be used in a practical field; this method does not require any expertise and skills. Because this is essentially a PCR-based method, the sensitivity is almost the same as that of the conventional PCR, and because the sequence of the target is confirmed by hybridization in MTH, the specificity is significantly higher compared to the other methods.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print