Introduction
Xanthomonas oryzae pv. oryzae (Xoo) is a Gram-negative bacteria that causes bacterial blight disease in rice (Niño-Liu et al., 2006). Xoo infection initiates from the leaf sheath and eventually spreads to mature leaves through the water flow under optimum temperature and high humidity conditions (Niño-Liu et al., 2006). Bacterial leaf blight disease caused by Xoo can result in a 20-50% yield loss in rice (Zhang and Wang, 2013). Therefore, analyzing the interaction between rice and Xoo could provide avenues for the genetic modification of crops against bacterial leaf blight disease (Gupta et al., 2015; Wang et al., 2017).
In the past decade, efforts have been made to elucidate the lifecycle, pathogenicity, and adaptation mechanisms of Xoo using various omics tools, including genomics, transcriptomics, and proteomics (Kim et al., 2016; Soto-Suarez et al., 2010; Wang et al., 2017; Zhang et al., 2013). Comparison of differentially expressed proteins among phytobacteria cultured under different conditions has been carried out by several research groups (Mehta and Yoko, 2001; Tahara et al., 2003; Villeth et al., 2009; Wang et al., 2017; Zhang and Wang, 2013). Xoo secretome analysis led to the identification of various hydrolytic enzymes and proteins involved in multiple functional categories, such as protein degradation and cell wall modification (Kazemi-Pour et al., 2004; Watt et al., 2005). Differentially modulated proteins were previously identified from the culture medium of X. axonopodis pv. citri, supplemented with the leaf extract of the host plant (Mehta and Yoko, 2001) or X. axonopodis pv. passiflorae (Tahara et al., 2003). A similar method was employed to investigate differentially modulated proteins of X. campestris pv. campestris (Villeth et al., 2009). Proteins of X. campestris pv. campestris cells were extracted and identified from susceptible plants and compared with those from a resistant plant (Villeth et al., 2009). Among these studies, enrichment of Calvin cycle-related proteins was detected, suggesting that regulation of energy metabolism and carbon fixation processes is essential for the interaction between host and pathogen.
Compared to a proteomic approach, a microarray-based high throughput approach is more widely used for genome-wide investigation of bacterial transcripts under in vitro and in planta growth conditions (Dugé de Bernonville et al., 2014; Zhou et al., 2011). However, microarray technology has limitations in the detection of weakly expressed genes. This is a major bottleneck, especially during in planta growth analysis of bacterial cells since the percentage of bacterial infected host cells is always less than the total host and microbe cell numbers. Therefore, to detect the transcriptional changes in Xoo under in planta growth conditions, two methods can be employed. Firstly, next-generation sequencing technology such as RNA-Seq has been developed to reduce inherent defects in microarray methods (Wang et al., 2009). RNA-Seq technology is a powerful method for high-throughput transcriptome analysis based on deliberate measurement of gene expression levels under given sample conditions. Especially, Illumina Genome Analyzer has been successfully employed to examine differential gene expression in pathosystems, including Xanthomonas axonopodis pv. glycines in soybean (Orshinsky et al., 2012), Sclerotinia homoeocarpa in bentgrass (Adhikari et al., 2012), and Pseudoperonos poracubensis in cucumber (Hinton et al., 2004). Secondly, enrichment of bacterial cells under in planta growth conditions could aid detection as well as purity of pathogen transcription (Tokuda et al., 2008). Thus, a combination of RNA-Seq and bacterial enrichment methods could increase our understanding of Xoo behavior under in planta infection conditions.
To date, there is no report on the transcriptome analysis of Xoo genes, obtained from in planta growth conditions, probably because of the difficulties in obtaining pure Xoo RNAs from in planta grown bacteria. Moreover, there is no information on the comparative transcriptome analysis of the Xoo genes grown under in vitro and in planta growth conditions. Alternatively, prior studies have attempted to monitor differentially expressed genes and proteins from bacteria in medium incorporated with host plant leaf extract in order to omit the RNA and protein extraction steps from in planta grown bacteria contaminated by host RNA and proteins, respectively (Mehta and Yoko, 2001; Tahara et al., 2003). However, few reports have provided suitable techniques to purely isolate RNA and proteins from in planta cultured bacteria cells for transcriptomic or proteomic analysis, respectively, during interactions between bacteria and the host plant (La et al., 2008; Wang et al., 2013). Recently, a highly efficient method for the isolation of in planta grown bacteria cells was reported in rice leaves using CaCl2 extraction buffer to purify bacterial samples from the debris of an infected host plant (Wang et al., 2017).
In this study, bacterial cells were harvested in adequate amounts from susceptible (Dongjin cv.) and resistant (Samgwang cv.) hosts using a CaCl2 method and subjected to RNA-Seq based transcriptomic analysis in order to gain deeper insights into in vitro and in planta expressed genes of Xoo during pathogenic interactions with rice. Illumina Genome Analyzer sequencer was employed to identify differentially expressed genes of Xoo grown under in vitro and in planta growth conditions.
Materials and Methods
Bacteria and plant growth conditions
Xoo strain K3 (KACC 10385/JH89011) was used in this study. Two rice cultivars (Oryza sativa L. cultivar Dongjin and Samgwang), which are compatible and incompatible with Xoo K3 strain, respectively, were used. Rice seeds were imbibed in sterilized water at 4°C for 2 days, germinated on moist tissue paper for 7 days, and transferred to soil in a growth chamber at 25°C (16/8 h day/light cycle, 70% relative humidity). Rice at the tillering stage was used for Xoo inoculation.
Xoo culture and infection of rice
Xoo strain was grown on Peptone sucrose agar (PSA) plate (1% w/v peptone, 1% w/v sucrose, 0.1% w/v glutamic acid, and 1.5% agar) at 28°C. For in vitro culture, Xoo cells were incubated in Peptone sucrose broth (PSB) medium and shaken at 200 rpm in the dark at 28°C for 3 days. Cultured cells were washed twice with distilled water and diluted to 108 cells mL−1 (OD600 = 0.1) with water containing 0.01% tween-20. Leaves from 50-day-old (at the tillering stage) rice cultivars were artificially infected with Xoo strains (108 cells mL−1) by clipping the leaf tips with sterile scissors. Infected rice plants were grown under normal light conditions (16 h light/ 8 h dark) for 5 or 10 days.
Xoo cell collection from in vitro culture medium and rice leaves
Cultured Xoo cells on PSB medium were collected by centrifugation at 12,000 g for 15 min and washed twice with distilled water. The in planta grown Xoo cells were collected using the CA extraction method (Wang et al., 2017). Briefly, infected rice leaves were detached, divided into 3 cm segments, immediately immersed in CA extraction buffer (200 mM CaCl2, 5 mM Na-acetate, pH 4.3) on ice, and shaken at 170 rpm for 1 h. Xoo cells, collected by filtering extraction buffer through filter paper, were harvested by centrifugation at 12,000 g for 15 min at 4°C. Cell pellet was frozen in liquid nitrogen and then used for RNA extraction.
RNA extraction and library preparation
Total RNA was extracted using a GeneAll Hybrid RTM Kit (GeneAll Biotechnology, Korea) according to the manufacturer’s instructions. RNA purity and concentration were assessed using an RNA Nano 6000 Assay Kit (Agilent Technologies, CA, USA) with the Bioanalyzer 2100 system (Agilent Technologies). For mRNA library construction and deep sequencing, RNA samples were prepared using a TruSeq RNA Sample Preparation Kit (Illumina, San Diego, CA, USA) according to the manufacturer’s instructions. Finally, the two libraries were sequenced using the Illumina HiSeq2000 platform in accordance with the manufacturer’s protocol (Illumina, Inc., San Diego, CA, USA). Construction of libraries and RNA sequencing were performed by the National Instrumentation Center for Environmental Management (NICEM) at Seoul National University (http://nicem.snu.ac.kr/). The quality control process was conducted to filter high-quality RNA reads using an NIPGR, Delhi, India (v2.3.3) (Zhao et al., 2011). Data normalization for differentially expressed analysis was calculated by RPKM (Reads Per Kilobase per Million reads) as described previously (Wagner et al., 2012).
GO and KEGG enrichment analysis
To analyze the functional properties of the identified Xoo genes, gene ontology (GO) enrichment analysis was carried out in the categories of biological processes and molecular function using the Universal Protein Resource online web-based toolkit (http://www.uniprot.org/) as described previously (Kim et al., 2011). Hierarchical clustering and Heatmap were generated using MA, USA (Orshinsky et al., 2012). Genome information resource was observed from KEGG (http://www.genome.jp/kegg/) and identified Xoo transcripts from susceptible and resistant hosts were mapped into the KEGG pathways using BLAST KOALA program (http://www.kegg.jp/blastkoala/) (Nakaya et al., 2013).
Prediction of putative secreted proteins
Different web-based prediction tools were employed to check the secretory gene products. Classical secreted proteins, derived from N-terminal signal peptides, and non-classical secreted proteins were predicted using SignalP (version 4.1) (http://www.cbs.dtu.dk/services/SignalP/) (Petersen et al., 2011) and SecretomeP (version 2.0) (http://www.cbs.dtu.dk/services/SecretomeP/), respectively (Bendtsen et al., 2005a). Bacterial specific twin-arginine signal peptide-containing proteins were predicted by TatP (version 1.0) (http://www.cbs.dtu.dk/services/TatP/) (Bendtsen et al., 2005b). Type III secretion effectors were predicted by the Effectors 4.0 online program (http://www.effectors.org/) (Jehl et al., 2011).
Semi-quantitative RT-PCR
To analyze expression levels of in planta Xoo genes, semi-quantitative real-time PCR was conducted using a Maxime RT PreMix Kit (iNtRON Biotechnology, Seongnam, Korea) according to the manufacturer’s instructions. Equal amount of purified RNA was used to synthesize cDNA, using a first strand cDNA synthesis kit (Sigma-Aldrich, Germany). PCR was performed with 30 cycles using gene-specific primers. The primer sequences used in this study are listed in Supplementary Table 1.
Results and Discussion
Differentially expressed genes during in planta growth
A total of 92,620,466 raw reads were identified from in vitro cultured Xoo; 94,542,634 and 104,550,144 raw reads were identified from in plant gown Xoo in susceptible and resistant hosts, respectively (Supplementary Table 2). Among raw reads from Xoo under in vitro conditions, more than 97.45% of bases had a q-value ≥ 20, whereas under in planta conditions, 97.92% (susceptible) and 97.71% (resistant) satisfied this criterion. GC contents were 53.64%, 53.75%, and 53.82% under in vitro as well as in planta susceptible and resistant conditions, respectively. There were 4,468,510,187 clean reads for Xoo under in vitro conditions as well as 5,235,181,135 and 4,697,020,133 clean reads under in planta conditions (susceptible and resistant), respectively (Supplementary Table 2, 3). Expression levels of genes were determined in accordance with the RPKM method (Wagner et al., 2012). Differentially expressed Xoo genes (DEGs) under in planta and in vitro conditions were selected based on criteria [q-value ≥ 0.001 and log2 (≥ 2 fold change)] using the MeV program (Supplementary Fig. 1). A total of 4,619 genes, including gene clusters with high (2784) and low (548) expression levels, in given sample sets, were identified (Supplementary Table 4). Among all DEGs, 81 Xoo genes were differentially expressed under susceptible and resistant interactions. Interestingly, over 10% of total genes were identified as transposable elements with approximately 25 kinds of transposase genes found in both the samples. Transposable elements (TEs) lead to chromosomal rearrangements such as deletion, inversion, duplication, and translocation (Hacker and Kaper, 2000). Xoo TEs were classified into six TE families: TE3, TE4, TE5, TE30, ISNCY, and TE630. In the Xoo genome, the most abundant TE was the TE5 family for which 117 copies were detected out of 207 total identified TEs. Many of these elements are located adjacent to strain-specific genes in regions with altered codon usage and G+C content. Of these, TEXo5 transposase was most abundant, followed by TEXo8 and TEXo3. However, expression of TEXo5 was higher in S than R cultivars. Several TEs and transposon sequences can be found in virulence-associated genes. Therefore, these elements might play important roles in horizontal gene transfer and internal modification of the genome. Furthermore, these TEs can convert pathogenic genes into non-pathogenic ones (Hacker and Kaper, 2000). These observations indicate that higher expression of TEs under in planta conditions could be a means of adaptation in the host plant.
Functional annotation and classification of identified genes
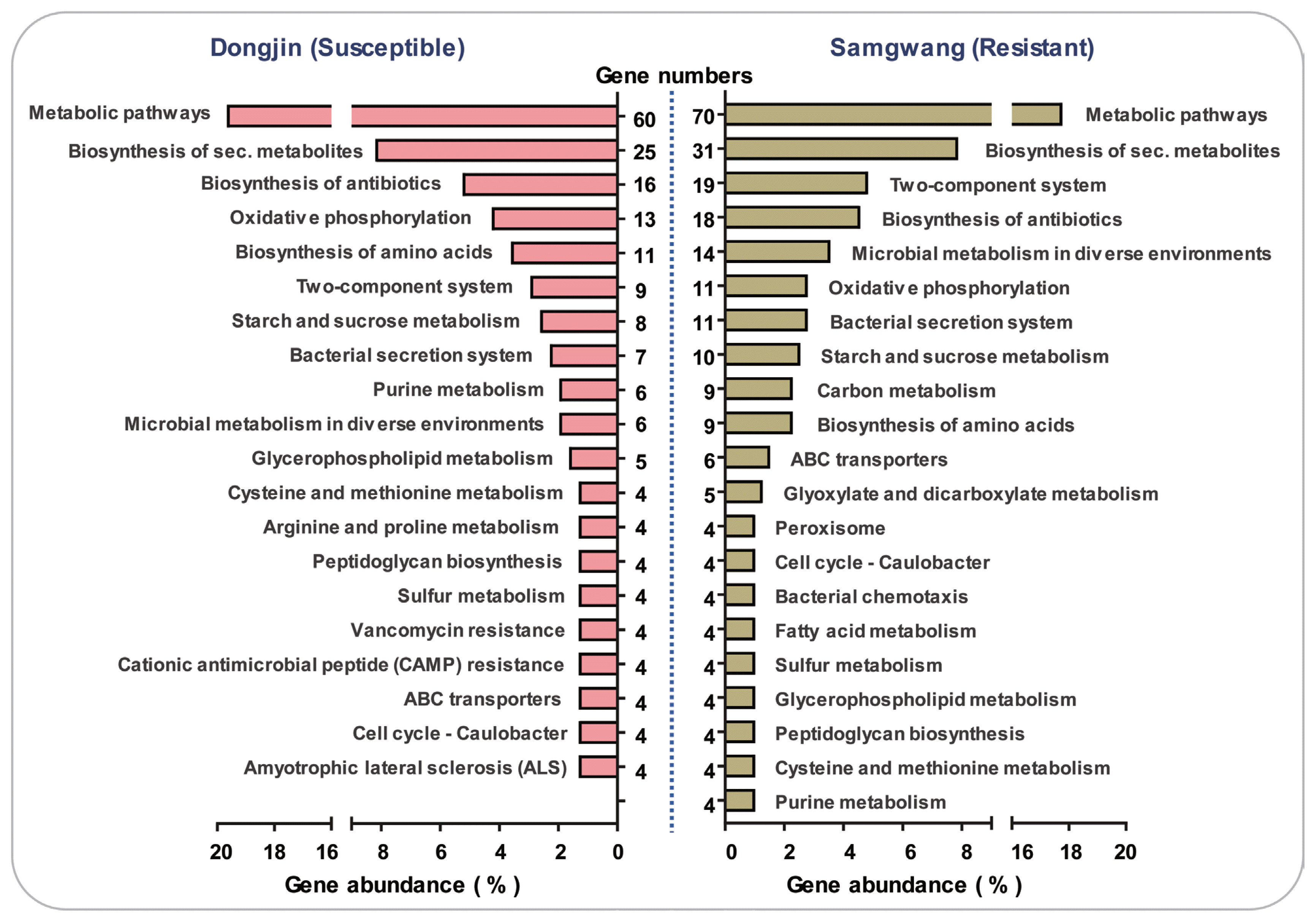
GO term enrichment analysis was carried out to determine the functional roles of DEGs between in planta and in vitro conditions (Fig. 1, Supplementary Table 5). A total of 1,619 Xoo genes from the susceptible host and 1,127 genes from the R host were functionally annotated into the molecular function and biological process categories (Fig. 1). In the molecular function category, up-regulated genes from the susceptible host were clustered into 27 categories. The largest category was ‘transporter activity’ (12.7%), followed by ‘cation binding activity’ (9.26%). In the case of biological processes, genes were divided into 14 categories of which the most abundant was ‘single-organism process’ (39.5%). Up-regulated Xoo genes from the resistant host were clustered into 13 groups, and most of these were related to ‘cation binding’ (23.68%) and ‘metal ion binding’ (18.42%) activities. For biological processes, the largest category was ‘carbohydrate metabolic process’ (32.2%). Organism processes, including cell proliferation, utilization of nutrition sources, and membrane organization, were highly detected in the susceptible host, whereas most of these were related to the carbohydrate metabolic process in the resistant host. In the molecular function category, genes related to cation binding activity were predominant in both susceptible and resistant interactions. Cation binding proteins play crucial roles in reactive oxygen species (ROS) stress as well as uptake of ammonium and metallic cations (such as copper, iron, and nickel) (Tam and Saier, 1993). It is well known that essential nutrients, such as iron, are required for the infection processes of pathogenic bacteria (Messenger and Barclay, 1983). In addition, the ability to scavenge these nutrients is required for survival and multiplication of pathogens (Hacker and Kaper, 2000). KEGG analysis revealed no major differences between susceptible and resistant hosts. In both the cases, ‘metabolic pathways’ and ‘biosynthesis of secondary metabolites’ were major pathways, and other parts such as ‘oxidative phosphorylation’ and ‘bacteria secretion system’ were detected (Fig. 2, Supplementary Table 6). However, a high percentage of genes involved in ‘microbial metabolism in diverse environments’ and ‘carbon metabolism’ pathways were observed in the resistant host. These results indicate that Xoo cells in the resistant host were under stress as compared to the susceptible host. Compared with in vitro cultured Xoo, in planta grown Xoo were more exposed to unfamiliar and nutrient-deficient conditions. During in planta growth, especially in the resistant host, genes related to stress and carbohydrate consumption were highly expressed. However, a considerable number of metabolic processes involved in reproduction and development were detected under susceptible conditions. Taken together, these results constitute detailed information on differentially up- and down-regulated Xoo genes between susceptible and resistant host-Xoo interactions.
Prediction of putative effectors from Xoo under in planta conditions
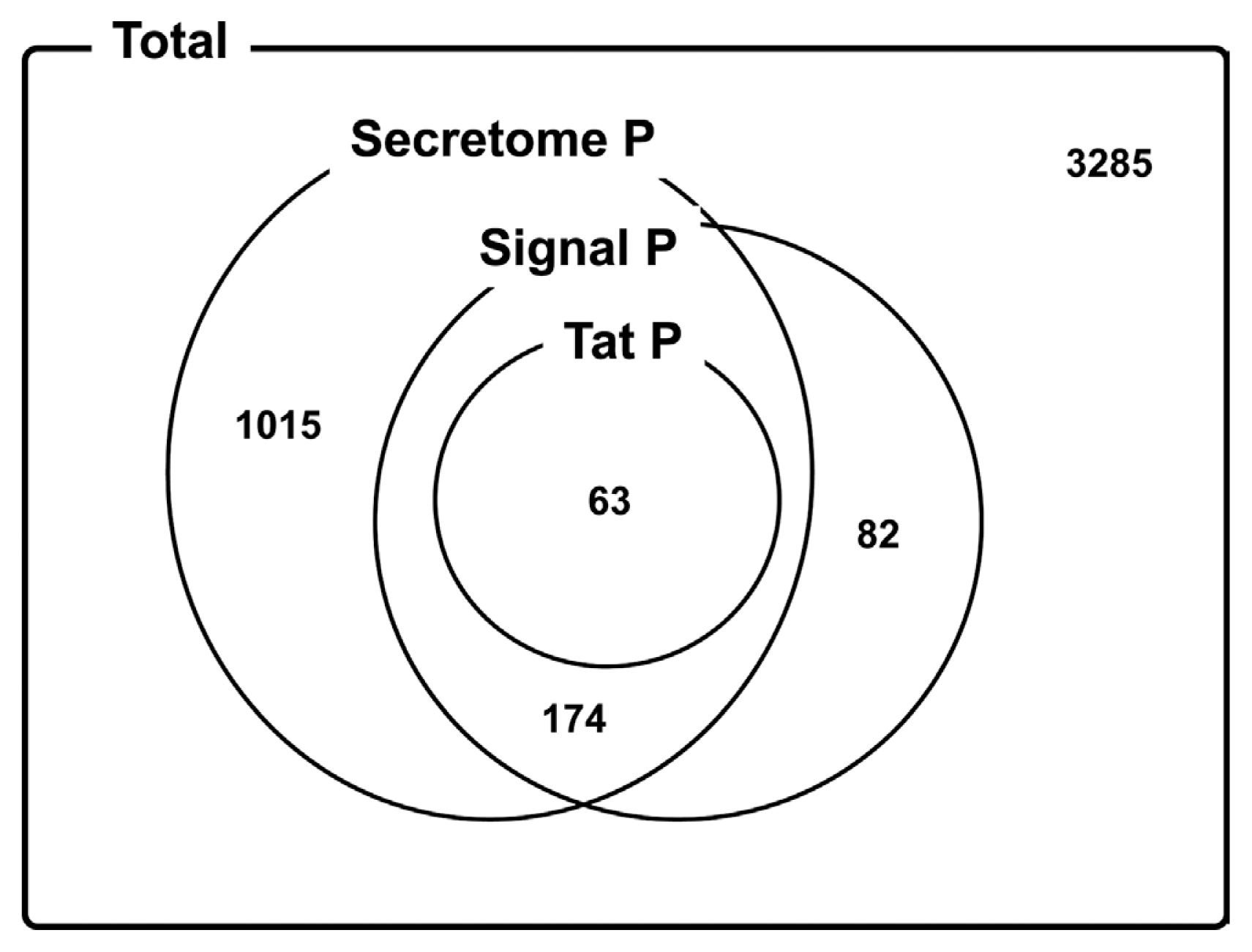
To determine putative secretory proteins and effectors from the identified genes, the SecretomeP, SignalP, and TatP programs were used. Of the identified genes, a total of 1,334 genes were found to have an N-terminal signal peptide and thus encode for the secretory proteins (Fig. 3). Of these secretory proteins, 184 putative type III effectors were predicted by a web-based analysis tool (Supplementary Table 7). The Hypersensitive reaction and pathogenicity (hrp), and avirulence genes (avr) accounted for a high percentage among others. Particularly, the AvrBs3 family, which is well known for its TAL (transcription activator-like) effector activity, was also detected in this study. TAL effectors act as transcription factors and influence host gene expression (Kay et al., 2007). Among these, candidate genes were sorted into their functional categories and combined together. Most of the genes related to nutrient uptake or transport showed similar expression patterns under in vitro and in planta growth conditions while genes related to protein degradation and modification and bacterial virulence were more highly accumulated in planta as compared to in vitro. Taken together, our results provide a deeper understanding of Xoo behavior during in vitro and in planta growth on resistant and susceptible hosts.
Validation of in planta expressed Xoo genes
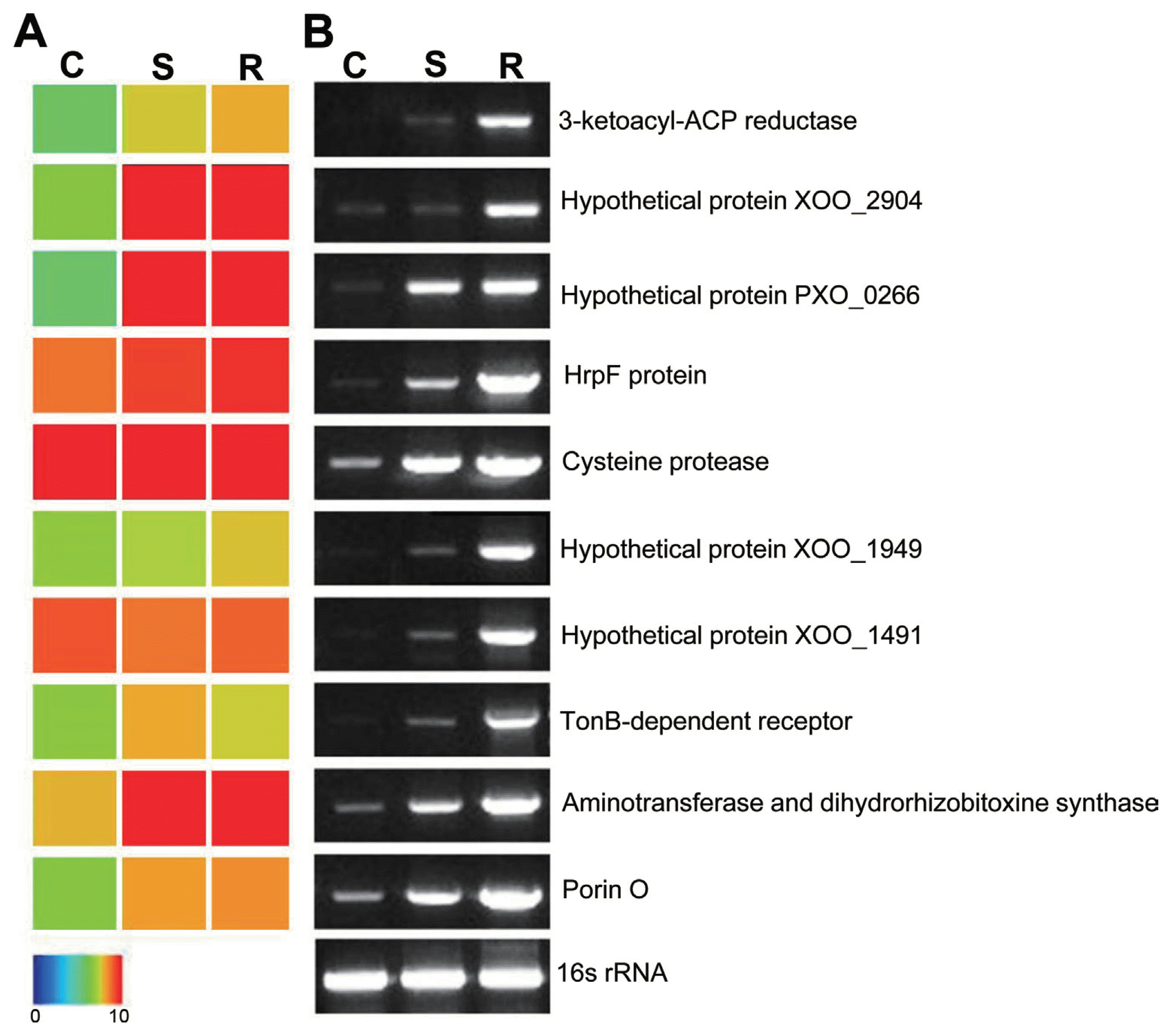
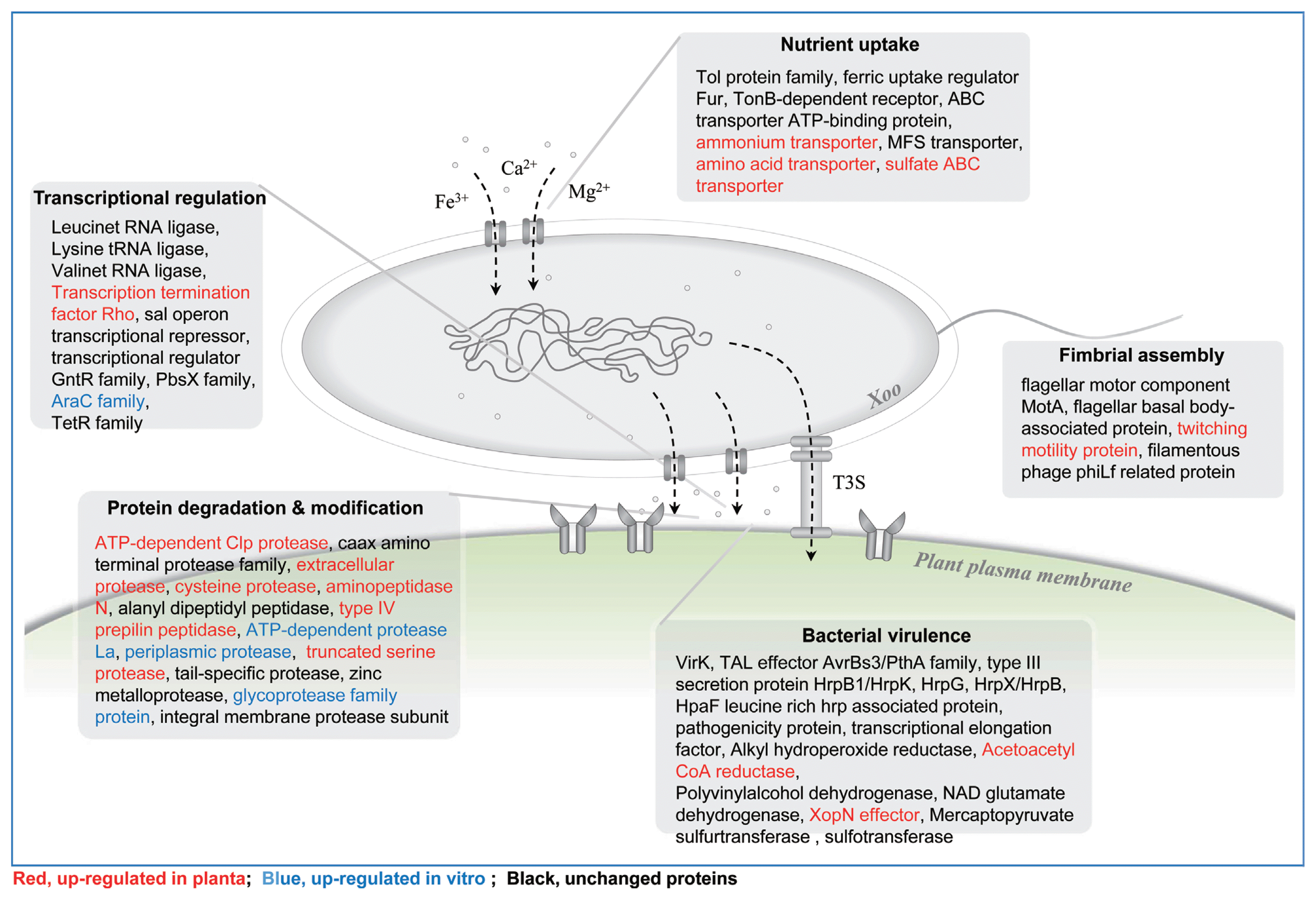
Transcriptome analysis showed that a total of 81 genes were highly up-regulated under in planta infection conditions as compared to in vitro culture conditions. To validate our transcriptome results, we randomly selected differentially expressed Xoo genes related to the pathogenicity (rtxA and HrpF), nutrition uptake (Porin-O and TonB), and fatty acid synthesis (FabG) and validated them by semi-quantitative RT-PCR (Fig. 4). ThertxA was reported to be involved in bacterial pathogenicity (Cirillo et al., 2001, 2002). HrpF is essential for the bacterial type III secretion system and is involved in pathogenicity (Rossier et al., 2000). Porin-O- and TonB- dependent receptors are related to the nutrition uptake for Xoo growth under in planta infection conditions (Blanvillain et al., 2007; Wang et al., 2002). FabG protein is known to be involved in bacterial type II fatty acid synthesis (Zhang et al., 2006). 16s rRNA primers were used as an internal control for normalization of cDNA concentration under each condition. Most genes were expressed under in planta infection conditions. In addition, genes related to motility such as flagella biosynthesis and chemotaxis were expressed. For several kinds of phytobacteria species, motility is an important factor for virulence (Josenhans and Suerbaum, 2002). Xoo secretes many extracellular enzymes during culture. In Xoo, cellulase and protease have been proposed to play important roles in bacterial virulence and nutrition (Ryan et al., 2011). Based on the results obtained here, an overview of Xoo candidate genes, which were differentially regulated under in planta and in vitro growth conditions, is provided (Fig. 5). A variety of genes involved in cell wall degradation were highly expressed under both in planta and in vitro growth conditions, including cellulase, xylanases, pectinases, proteases, xylosidases, and polygalacturonases.
Previously, comparative transcriptome analysis was performed on different Xoo strains exhibiting differential virulence in rice during in vitro culture in minimal medium (Zhang et al., 2013). Genes related to exopolysaccharide (EPS) synthesis, Hrp proteins, and type III effectors, including Xanthomonas outer protein (Xop) effectors and transcription activator-like (TAL) effectors, were differentially expressed between Xoo strains, suggesting these proteins may affect bacterial pathogenicity in plants. Later, Kim et al. (2016) performed transcriptome analysis of Xoo in response to rice leaf extracts under in vitro culture conditions. Leaf extracts were shown to induce rapid induction of genes related to ion transport, metabolism, cell motility, pathogenicity, chemotaxis, flagella biosynthesis, and cyclic-di-GMP control. However, the immune related genes or metabolites were induced upon pathogen infection, and these immune outputs play an important role in anti-pathogen processes (Jones and Dangl, 2006; Withers and Dong, 2017). Therefore, investigation of transcriptome or proteome changes during in planta infection condition, which could not be monitored via in vitro conditions, will provide an in-depth understanding of plant-microbe interaction. Therefore, a microarray-based transcriptome analysis of Xoo during in planta infection condition was carried out in a susceptible rice species (Soto-Suárez et al., 2010). Differentially expressed genes were related to metabolism, secretion, and transport, pathogen adherence to plant tissues, plant cell-wall degradation, IS elements, and virulence. In a combination of that information, here, a comparative transcriptome analysis of Xoo under in vitro and in planta growth conditions was performed for the first time.
In summary, a total of 4,619 Xoo genes were identified under in planta and in vitro growth conditions. Of these, some of the interesting candidates identified included TAL effector or protein synthesis related genes, EF-Tu, and transposase and these were up-regulated during in planta Xoo growth conditions. As these proteins are highly expressed during in planta growth conditions only, these can be directly linked to the Xoo pathogenicity. In addition to these, 184 effector candidate genes were also identified including 10 candidate effector genes that were especially expressed under in planta conditions. These genes encodes for HrpF, proteases, and TonB. Taken together, list of genes and effectors provided here can serve as resource for further validation and their utilization to genetically modify the Xoo genome to reduce its pathogenicity.

 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print