Comparison of Microbial Community Structure in Kiwifruit Pollens
Article information
Abstract
Flowers of kiwifruit are morphologically hermaphroditic and survivable binucleate pollen is produced by the male flowers. In this study, we investigated microbial diversity in kiwifruit pollens by analyzing amplicon sequences of 16S rRNA. Four pollen samples were collected: ‘NZ’ was imported from New Zealand, ‘CN’ from China in year of 2014, respectively. ‘KR13’ and ‘KR14’ were collected in 2013′ and 2014′ in South Korea. Most of the identified bacterial phyla in the four different pollens were Proteobacteria, Actinobacteria and Firmicutes. However, the imported and the domestic pollen samples showed different aspects of microbial community structures. The domestic pollens had more diverse in diversity than the imported samples. Among top 20 OTUs, Pseudomonas spp. was the most dominant specie. Interestingly, a bacterial pathogen of kiwifruit canker, Pseudomonas syringae pv. actinidiae was detected in ‘NZ’ by the specific PCR. This study provides insights microbial distribution and community structure information in kiwifruit pollen.
Plant-associated microbes are fundamental determinant of plant health and productivity (Berendsen et al., 2012). Microbes living on surfaces of plant are called epiphytes and living in inner tissues are endophytes. Habitat of plant-associated microbes are mainly divided into phyllosphere which is the aerial surfaces of fresh plant on the ground and rhizosphere which is the attached soil of root surfaces (Johnston-Monje et al., 2016). These plant microbiome makes the host to enhance nutrient accumulation from the soil, tolerate against abiotic stresses, produce phytohormones and protect from pathogens (Bulgarelli et al., 2013). Accordingly, it is crucial to understand the plant microbiome that how they coexist and what they do ecosystem. In the laboratory, microbes are separated on culture media and identified with biochemical and morphological methods (Yang and Crowley, 2000). However, the total environmental DNA sequence data suggested that less than 1% in a gram of soil microbes can be cultured. As the ‘Pyrosequencing’ or ‘Metagenomics’ emerges, it can explore microbial community and diversity research without culturing microbes (Gilbert et al., 2010). Therefore, the microbial studies using the culture-independent methods have recently provided much more ecological information of phyllosphere microbiology than previously studied (Lopez-Velasco et al., 2013). The plant rhizosphere contains the vast microbial diversity that colonize on the roots. One gram of soils contains about 2 × 109 CFU (Daniel, 2005). The majority of bacteria in the rhizosphere habitats belonged to Proteobacteria, Acidobacteria, Actinobacteria and Bacteroidetes. Microbial community composition differs depending on the plant species, the soil type and the cultural practices. Plant organic matter such as root exudates, epidermal cells and mucilage also influence the microbial community in rhizosphere (Coats et al., 2014). Compared with bulk soil, the microbial biomass and activity in rhizosphere are enhanced as a result of compounds exudation by the root (Sørensen, 1997). Likewise, such studies focused on rhizosphere have been widely reported. However, phyllosphere microbiome has been less studied then rhizosphere one (Vorholt, 2012). Bacteria have often distributed in numbers averaging 106 to 107 cfu/cm2 on leaves. Proteobacteria containing α-, β- and γ-proteobacteria are known as the dominant bacterial group of the phyllosphere with Firmicutes, Acidobacteria, Actinobacteria and Cyanobacteria (Whipps et al., 2008). Most studies about phyllosphere microbial community has focused on leaves but pollen-associated microbes are poorly investigated. Pollen-associated microbial communities are different by pollination types such as insect-pollinated plants and wind-pollinated ones or species-specific in same environmental conditions (Ambika Manirajan et al., 2016).
Actinidia spp. (Kiwifruit) is an economically important crop widely cultivated in many countries. It is dioecious fruit tree divided into opposite sex: male and female flower. These flowers are morphologically hermaphroditic, however, survivable binucleate pollen is produced by the male flowers only (Fraser et al., 2009). The kiwifruit bacterial canker caused by Pseudomonas syringae pv. actinidiae (PSA) is the severe problem in the global kiwifruit industry (Kim et al., 2017). The symptoms of bacterial canker appear brown spots surrounded by a yellow halo on the leaves, milky bacterial exudates from the many organs (twigs, vines, leaves and flower) and typical canker on the trunks (Kim et al., 2017). Pollen contaminated by the PSA is a serious vector to spread the canker pathogen (Chapman et al., 2011). Overwintering PSA can disperse to host via pollen in the disease cycle (Donati et al., 2014). Artificial pollination with the PSA contamination pollen obviously spread and transfer the pathogen in orchard and transfer the pathogen from infected orchard to non-infected orchard (Tontou et al., 2014). Therefore, understanding of the microbial community structures of the pollen is necessary to implement the management strategies of kiwifruit bacterial canker.
In this study, four kiwifruit pollen samples were collected from several orchards in New Zealand, China and South Korea. Two pollen samples, New Zealand and China were imported in year of 2014 and two domestic pollen samples were collected from a kiwifruit orchard in Sacheon, Korea in year of 2013 and 2014, respectively. One gram of the pollen was added to a test-tube containing 9 ml of 1X Phosphate buffered saline (PBS; 8 g NaCl, 0.2 g KCl, 1.44 g Na2HPO4, 0.24 g KH2PO4, bring total volume to 1l with ddH2O, pH 7.4). To lyse the cellular organelles and extract DNA, the samples were sonicated with frequencies of 35 kHz for ten minutes using ultrasonic bath (Bandelin electronic GmbH and Co., Berlin, Germany). Suspensions were transferred to 1.5 ml tube, centrifuged at 13,000 rpm for 1 min then the supernatant was discarded. The process was repeated 3 times. Total DNA were extracted using DNA prep kit (Solgent Co., Daejeon, Korea) divided into gram-negative and gram-positive. In the case of gram-positive bacteria, the pellets were resuspended in 300 μl of R1 solution, added 2 μl of Lysozyme (100 mg/ml). The tubes were incubated at 37°C for 1 h, centrifuged at 10,000 rpm for 1 min and the supernatant was discarded. From here, experimental procedures were performed as same both gram-positive and gram-negative bacteria. SGD1 solution (350 μl) and 5 μl of protenase K (20 mg/ml) were added, mixed using vortex for 1 min and incubated at 65°C for 10 min. After cooling the mixtures thoroughly at room temperature for 5 min, 400 μl of SGD2 solution was added, mixed then centrifuged at 10,000 rpm for 5 min. During centrifuged, spin column was installed to 2-ml collection tube. SuperBinder™ Solution (100 μl) was added to the spin column, centrifuged at 10,000 rpm for 30 sec and the pass-through was discarded. Spin column was transferred to a new 2-ml collection tube. WB (500 μl, 80% Ethanol) was added to spin column and centrifuged at 10,000 rpm for 30 sec. The pass-through was discarded, in the same way, DNA was washed with 500 μl of WB (80% Ethanol) once again. The empty spin column was centrifuged at 10,000 rpm for 2 min to eliminate the WB completely and transferred to a new 1.5 ml tube. DNA Hydration Solution (50 μl) was added, incubated at room temperature for 1 min and centrifuged at 10,000 rpm for 2 min. Spin column was eliminated and final DNA were stored in a new 1.5 ml tube at -80°C.
Bacterial 16S rRNA regions were amplified by forward primer 27F (10 pmol): 5′-GAGTTTGATCMTGGCTCAG-3′ and the reverse primer 518R (10 pmol): 5′ WTTACCGCGGCTGCTGG 3′ (Kwon et al., 2010). The volume of PCR products was adjusted to total volume 20 μl with 13.8 μl of distilled water, 100 ng of template DNA, 2 μl of 10X reaction buffer, 1 μl of 10 mM dNTP, 1 μl of respective primers and 5 unit of Taq DNA polymerase (Bioneer Co., Daejeon, Korea). Reactions were performed using 5 min of denaturation step at 95°C, 30 cycles consisting of 30 sec at 95°C, 30 sec at 55°C and 30 sec at 72°C, followed by 5 min of final extension at 72°C (Rondon et al., 2000). PCR products containing the amplified DNA fragments were checked their size by electrophoresis on 1% agarose gel containing ethidium bromide in 1% Tris-acetate EDTA (TAE) buffer. The PCR products were purified using the Expin™ gel SV kit (Geneall biotech Co., Seoul, Korea). Pyrosequencing was performed using the Roche GS FLX software (v 3.0) by a 454 GS-FLX titanium system (Roche, Pleasanton, CA, USA). The reads were segregated from each sample using tag (Barcode) sequences and matched the initial and final bases of the reads to the known barcode sequences used in the preparation of the libraries. The sequenced data were analyzed using the MG-RAST metagenomics analysis server (version 4.0, http://v3-web.metagenomics.anl.gov). Operational taxonomic units (OTUs) were determined to compare the bacterial richness among the samples. Total read count was 100,640 of which ‘NZ’ was 29,078, ‘CN’ was 24,636, ‘KR13’ was 24,588 and ‘KR14’ was 22,338 with 97% similarity. Ambiguous counts, short sequences lower than 174, chimeric counts and other all noises were filtered. Most of the identified bacterial phyla in the 4 different pollens were Proteobacteria, Actinobacteria and Firmicutes (Fig. 1A). These three phyla have major probiotic microbes, for example, alpha and beta proteobacteria, Streptomyces spp. in Actinobacteria and Bacillus spp. in Firmicutes (Noh et al., 2017). Proteobacteria was identified at the highest rate more than 60% in all samples. Compared the imported and domestic pollens, they had different microbe community (Fig. 1B). In the imported samples, Proteobacteria was approximately 80%. In the domestic samples, Firmicutes or Actinobacteria ranged about 15% differently and bacterial diversity also changed after a year from 13′ to 14′. In KR13, Firmicutes was more abundant than Actinobacteria, but there were flipped in KR14, Actinobacteria became second most abundant bacteria and followed Firmicutes in KR14. These findings may cause by limited technical replication of samples.
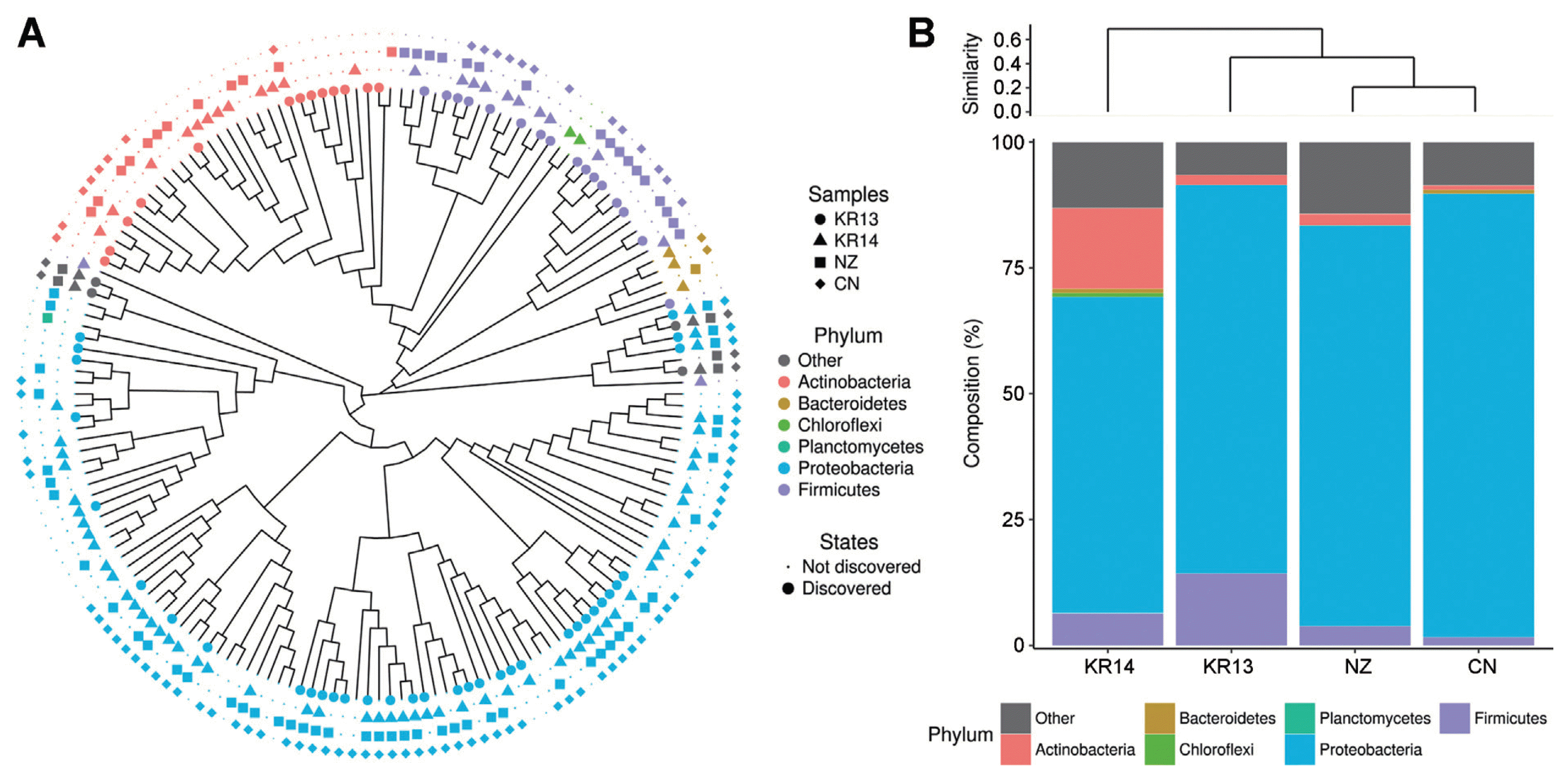
Proportional bacterial communities of different phyla in the kiwifruit pollens. (A) Total bacterial communities of different phyla showing all samples. (B) Respective microbe abundant ratios. Imported samples indicated that of New Zealand ‘NZ’ and of China ‘CN’ and domestic samples indicated that of South Korea ‘KR13’ and ‘KR14’.
The number of OTUs was that ‘NZ’ was 86, ‘CN’ was 99 ‘KR13’ was 70 and ‘KR14’ was 92. Although total bacterial OTUs can be ranged to 10000 in one gram of soil, the total OTUs were too low in the same amount of pollen (Roesch et al., 2007). Venn diagram showed that four different pollens shared distinct core microbes only 31 of 172 OTUs as 18% (Fig. 2). The core OTUs occupied with 99.8% of total read counts and 78.6% was Proteobacteria, 3.5% was Actinobacteria and 4.4% was Firmicutes. It can be considered that most of the microbes of the kiwifruit pollen belong to Proteobacteria. However, on OTUs, there were many unshared OTUs 17 of KR13, 25 of KR14, 22 of NZ and 15 of CN. In KR13 and KR14, although they had been from same flower, OTUs also have changed. It can indicate that the microbial structures of pollen can be changed by abiotic factors such as temperature, wind, wind and humidity or biotic factors such as bees and other plants (Obersteiner et al., 2016). When their similarities were also shown in Principal coordinate analysis (PCoA), ‘NZ’ and ‘CN’ had a correlation but ‘KR13’ and ‘KR14’ became less relevant as they had changed the bacterial diversity (data not shown). Top 20 OTUs more abundant in the imported than the domestic pollens were compared by the abundant OTU proportions (Table 1). The top 20 OTUs belonged to 4 phyla of Proteobacteria, Actinobacteria, Firmicutes and Bacteroidetes and 40% of them were the members of uncultured bacterium taxa. High OTUs belonged to Proteobacteria and Pseudomonas trivialis, P. putida and other Pseudomonas spp. were relatively high read numbers among the Pseudomonadaceae. P. trivialis is reported that it was isolated from the phyllosphere of grasses (Behrendt et al., 2003). P. putida is reported that it is antagonistic to soil-borne fungal pathogens such as Verticillium dahliae causing disease in strawberry, potato and oilseed rape (Berg et al., 2002). It is also known to play an important role in suppressing the apple replant disease caused by Cylindrocarpon destructans, Pythium ultimum and Rhizoctonia solani (Gu and Mazzola, 2003). In the case of top 20 OTUs more abundant in the domestic pollens than the imported pollens with the read number, likewise, 30% was identified as uncultured bacterium (Table 2). Most of the strains were belonged to Proteobacteria and the rest phyla were Actinobacteria and Firmicutes. P. fluorescens known as the antibiotics producer (Maurhofer et al., 1995), which suppressed fungal diseases caused by soil-borne pathogens, showed the highest proportion of 44.16% in ‘KR13’. Although the Proteobacteria was occupied much more in the imported samples than domestic samples, top 20 OTU portions of Proteobacteria were more occupied in the domestic samples when compared with number of OTU. Enterobacteriaceae was detected as the second most abundant OTU in both the imported and the domestic pollen samples. In the imported pollen, 20.09% of Enterobacteriaceae was identified as uncultured bacterium and 5.85% was Erwinia aphidocola, which is known as a pathogenic bacterium in several plants (Luo et al., 2018; Marín et al., 2011). In contrast, in the domestic pollen most of Enterobacteriaceae was assigned as Pantoea spp.
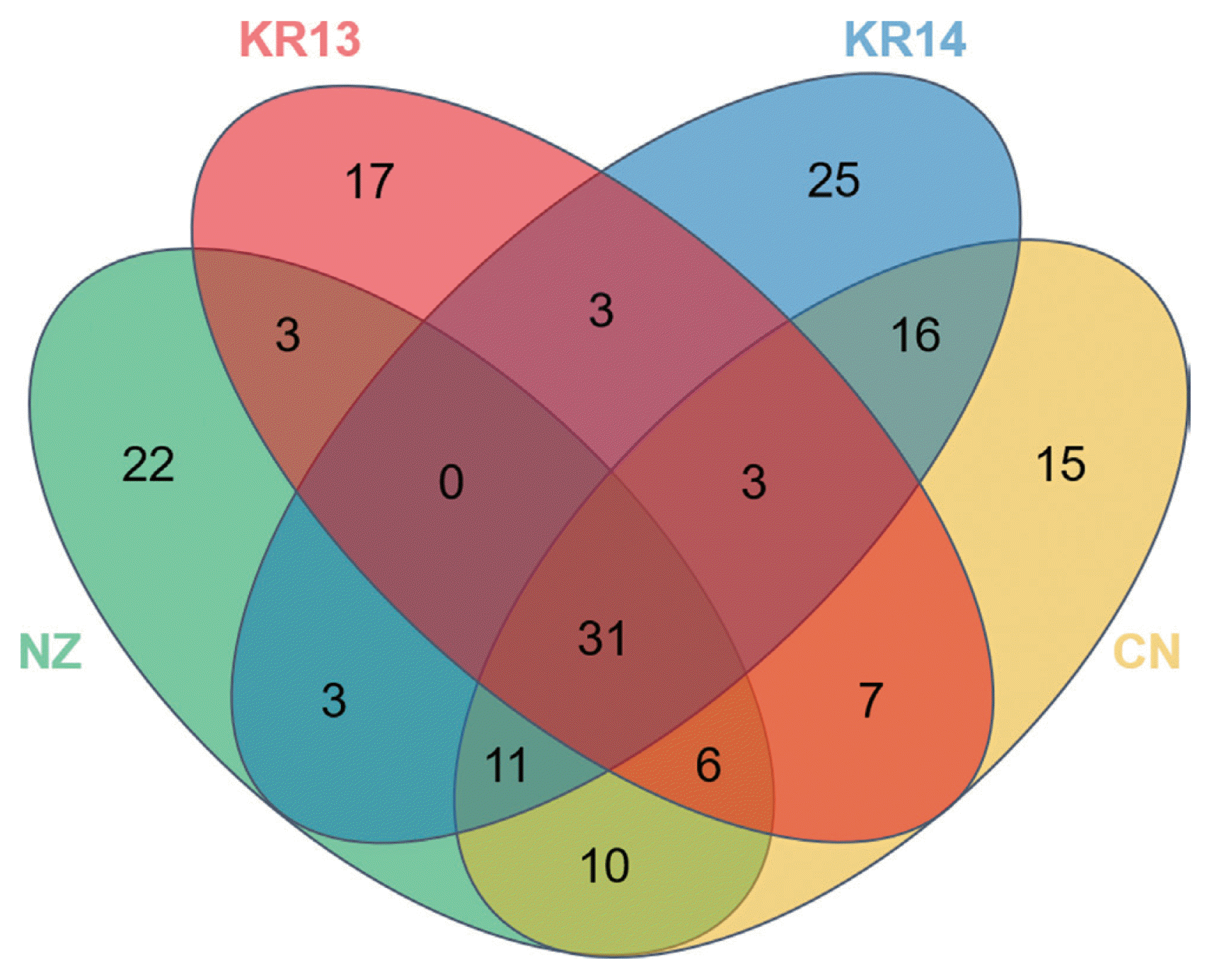
Venn diagram showing the shared OTUs in the four pollen libraries. CN: Chinese pollen, NZ: New Zealand pollen, KR13: Korean pollen of 13′, KR14: Korean pollen of 14′.

Top20 OTUs more abundant in the imported pollens than the domestic pollens

Top20 OTUs more abundant in the domestic pollens than the imported pollens
Although, the most abundant phyla were Proteobacteria, the pyrosequencing failed to detect PSA directly. In general, size of amplicon for pyrosequence is relatively short (near 300 bp) to identify in genus or species level of bacteria. Therefore, we employed PCR protocol with sensitive and specific primers to PSA. The PCR reaction was conducted using the DNA of the four pollen samples and an identified PSA strain (positive control). The primer pair was forward primer PsaF1 (10 pmol): 5′-TTTTGCTTTGCACACCCGATTTT-3′ and the reverse primer PsaR2 (10 pmol): 5′ CACGCACCCTTCAATCAGGATG 3′ (Rees-George et al., 2010). The region of 280 bp was amplified by PsaF1/PsaR2 primers. The volume of PCR products was same with the 16S rRNA genes amplification. Reactions were performed using 2 min of denaturation step at 95°C, 30 cycles consisting of 15 s at 95°C, 30 s at 55°C and 90 s at 72°C, followed by 5 min of extension at 72°C. Electrophoresis was performed to identify their presence as above. PSA was detected by the PCR only in ‘NZ’ (Fig. 3). In ‘NZ’, Pseudomonas spp. was the most identified among the samples as 61.64% and PSA was detected by specific PCR. The K13 sample showed a faint PCR amplicon. The result suggested that K13 pollen may be also contaminated by PSA. However, in this study we could not make final decision due to low intensity of the band. Therefore, in further study we should develop more sensitive and specific protocol to detect PSA such as qRT-PCR or LAMP-PCR.

Detection of PSA using the PSA detection primers, PsaF1/PsaR2 was used to detect the Pseudomonas syringae pv. actinidiae. 1% agarose gel was used to confirm the PSA in gel electrophoresis. M: 100 bp DNA ladder, CN: Chinese pollen, NZ: New Zealand pollen, KR13: Korean pollen of 13′, KR14: Korean pollen of 14′, CK: genomic DNA of PSA (positive control).
In conclusion, we provide the first insight on microbial community of the kiwifruit pollens. When the pyrosequencing of the pollen samples was conducted, the imported pollens had simple microbial diversity than the domestic samples. It is considered that there were differences by regions or change of microbial communities of the imported samples. The microbial communities were also changed from ‘KR13’ to ‘KR14’. It may be a natural change caused by numerous factors over time. Microbial information about pollen is not general but these studies have to performed for mismanagement of pathogen by pollen. Therefore, we suggest that the ecological information of kiwifruit pollens and detection of PSA are crucial to understanding and management of kiwifruit bacterial canker disease.
Acknowledgments
This research was supported by the ‘Cooperative Research Program for Agriculture Science & Technology Development (PJ010904)’ from the RDA, Republic of Korea.