Loop-mediated Isothermal Amplification assay for Detection of Candidatus Liberibacter Asiaticus, a Causal Agent of Citrus Huanglongbing
Article information
Abstract
Huanglongbing (HLB, Citrus greening disease) is one of the most devastating diseases that threaten citrus production worldwide. Although HLB presents systemically, low titer and uneven distribution of these bacteria within infected plants can make reliable detection difficult. It was known loop-mediated isothermal amplification (LAMP) method has the advantages of being highly specific, rapid, efficient, and laborsaving for detection of plant pathogens. We developed a new LAMP method targeting gene contained tandem repeat for more rapid and sensitive detection of Candidatus Liberibacter asiaticus (CLas), putative causal agent of the citrus huanglongbing. This new LAMP method was 10 folds more sensitive than conventional PCR in detecting the HLB pathogen and similar to that of real-time PCR in visual detection assay by adding SYBR Green I to mixture and 1% agarose gel electrophoresis. Positive reactions were achieved in reaction temperature 57, 60 and 62°C but not 65°C. Although this LAMP method was not more sensitive than real-time PCR, it does not require a thermocycler for amplification or agarose gel electrophoresis for resolution. Thus, we expect that this LAMP method shows strong promise as a reliable, rapid, and cost-effective method of detecting the CLas in citrus and can be applied for rapid diagnosis is needed.
Body
Huanglongbing (HLB, Citrus greening disease) is one of the most devastating diseases that threaten citrus production worldwide except Korea, Europe, Australia and New Zealand (CABI, 2018). HLB is caused by three fastidious α-Proteobacteria species of Candidatus Liberibacter, namely Candidatus Liberibacter asiaticus (CLas), Candidatus Liberibacter americanus (CLam) and Candidatus Liberibacter africanus (CLaf) (Gottwald, 2010). HLB-associated bacterium, CLas, is phloem-inhabiting, gram-negative and causes phloem collapse and phloem plugging, resulting in impairment of phloem transport of citrus. Consequently, carbohydrates partitioning in diseased plant lead to metabolism imbalance and tree decline (Etxeberria et al., 2009; Fan et al., 2013; Koh et al., 2012).
The general leaf symptoms of HLB are blotchy mottle, yellow vein and cork (Bové, 2006; Gottwald, 2010). The symptoms are similar to micro-element deficiencies, such as zinc, so it is difficult to diagnose with the naked eye. Although it has not been reported in Korea, it is necessary to establish a method that can diagnose HLB quickly and accurately before it invades and spreads in Korea. Many methods to diagnose HLB infection have been developed including iodine reaction based on the elevated starch accumulation in HLB-diseased citrus leaves (Takushi et al., 2007), serology using enzyme-linked immune-sorbent assays (ELISA) with monoclonal antibodies (Villechanoux et al., 1990), and symptomatology by observation midribs with leaf mottle using electron microscopy (EM). Molecular detection methods include DNA–DNA hybridization (Bové, 2006), conventional PCR used with 16S rDNA-based primers of three Ca. Liberibacter spp (Jagoueix et al., 1996; Nageswara-Rao et al., 2013; Teixeira et al., 2005), real-time PCR targeting the 16S rDNA-region (Bové, 2006; Kim and Wang, 2009; Li et al., 2006; Pietersen et al., 2010) as well as multiple genetic loci such as hyvI (LasAI) and hyvII (LasAII) in CLas prophage genes (Morgan et al., 2012; Zhou et al., 2011), and a combination of real-time PCR and nested PCR in a single tube to detect the elongation factor Ts gene (Lin et al., 2010).
Loop-mediated isothermal amplification (LAMP) can amplify target DNA under isothermal temperatures (60–65°C) and, requires Bst polymerase with strand-displacement activity and a set of four or six primers (Nagamine et al., 2002). LAMP has the advantage of being highly specific, rapid, efficient, and labor-saving. In contrast to both conventional PCR and real-time PCR, a precision thermocycler is not needed because LAMP is performed at a single temperature. Recently, LAMP was developed for the detection of CLas (Okuda et al., 2005; Rigano et al., 2014), Ca. Liberibacter solanacearum (Ravindran et al., 2012), Erwinia amylovora (Temple and Johnson, 2011), Tylenchulus semipenetrans (Song et al., 2017) and Wheat Streak Mosaic Virus (Lee et al., 2015). Although the LAMP method can be less sensitive than real-time PCR, it has the advantage of no need for a thermocycler for amplification or agarose gel electrophoresis for resolution. In this study, we present a newly developed LAMP method by using primer set targeting gene contained tandem repeat for reliable, rapid, and cost-effective method of detecting the CLas in citrus and can be applied for rapid diagnosis is needed.
Materials and Methods
Plant materials and genomic DNA extraction
Diseased leaves were collected from HLB infected grapefruit trees in the Indian River Research and Education Center (IRREC, Fort Pierce, FL) and healthy grapefruit leaves were sampled in IRREC Green house.
Total genomic DNA was extracted using a modified method of the Qiagen DNeasy Plant mini kit protocol (Qiagen, Germantown MD) reported previously (Hoffman et al., 2013). Two hundred milligrams of finely chopped midribs from leaf samples were put in a sterile 2 ml tube with silicone-carbide shards and, 2.3-mm chrome-steel beads, then then 800 ul AP1 extraction buffer and 80 ul of 20% SDS were added. Samples were homogenized with a Fast Prep-24 homogenizer (MP Biomedical, Solon, OH) at a speed of 6.5 for 100s. Eight microliters of RNase A was added and samples were incubated for 30 min at 65°C. Then 400 μl of AP2 buffer was added and samples were put on ice for 5 min. All subsequent processes were the same as the standard protocol. DNA samples were eluted in 100 ul of elution buffer and stored at −30°C.
Primer design for LAMP
Whole genome sequences of CLas strains, psy62, gxpsy and Ishi-1 have been completed (Duan et al., 2009; Lin et al., 2013; Katoh et al., 2014), but the LasAI and LasAII genes sequences of CLas isolates from Florida, China, Thailand, Philippines, India and Brazil, have also been reported previously (Puttamuk, 2014). For effective detection of CLas pathogens including these isolates, the LasAI gene sequences of these isolates were collected from the National Center for Biotechnology Information, aligned using Bio-Edit software, and it was determined that an area consisting of 269 bp from the start codon was highly conserved among these isolates (Fig. 1). A set of CLas-specific LAMP primers, F3, B3, FIP and BIP was designed based on this 269 bp length using PrimerExplorer software (http://primerexplorer.jp/e) (Table 1).
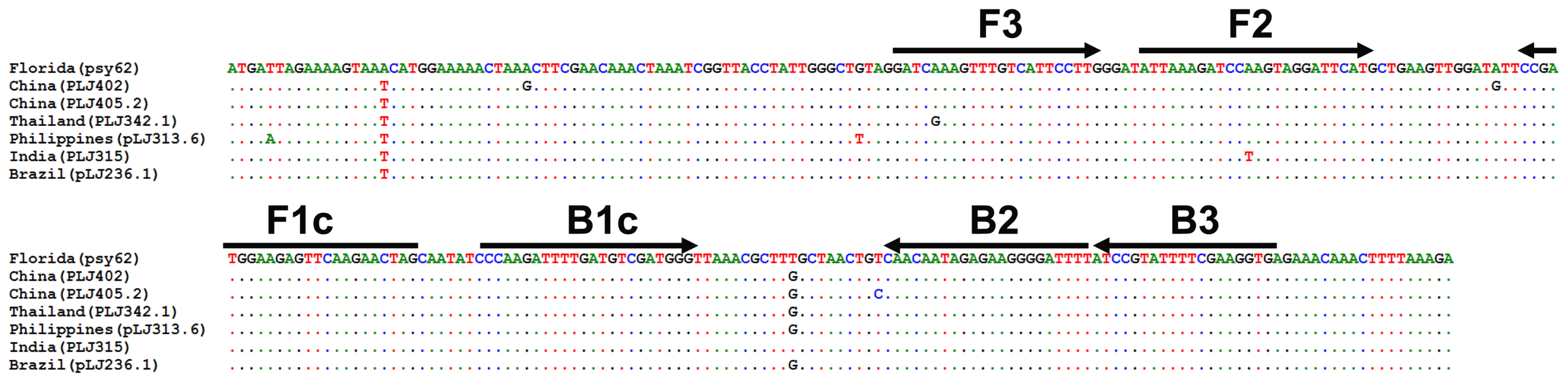
Alignment of partial of LasAI sequences of isolates from Florida, China, Thailand, Philippines India and Brazil, and primers designed for HLB loop-mediated isothermal amplification (LAMP). Length of 269 bp from start codon was used for primer design for LAMP PCR. Black arrows are indicated as primer target sequences and extension directions. The FIP and BIP primers consist of F1c plus F2 and B1c plus B2, respectively.

Sequence of primers/probe used in this study
LAMP reaction
LAMP reaction mixtures prepared in a final volume of 20 μl containing 1 μl template DNA, 2.0 μM of FIP and BIP primers, 0.5 μM F3 and B3 primers, 1 mM of dNTPs, 8 U of Bst DNA polymerase (New England Biolabs, Ipswich, MA, USA), 1× ThermoPol reaction buffer (20 mM Tris-HCl, 10 mM KCl, 2 mM of MgSO4, 10 mM (NH4)2SO4, 0.1% Triton X-100, pH 8.8). After pretreating for 10 min at 95°C and 1 min at 4°C, the reaction mixtures were reacted for 1hour at four different reaction temperatures (57, 60, 62 and 65°C) and terminated for 5 min at 85°C.
Detection of LAMP products
After completion of the reaction, the LAMP amplification results were analyzed visually by adding 2 μl of a 1:10 dilution of SYBR Green I (10,000x, Invitrogen, Grand Island, NY, USA) and reactions that turned green were scored as positive and those that remained brown were scored as negative. Additionally, the amplified LAMP products were also analyzed by 1% agarose gel electrophoresis, stained with ethidium bromide and then photographed under a UV light using Gel Doc™ XR+ Gel Documentation System (Bio-rad Labolatories, Inc. Herules, CA).
Sequence analysis of LAMP product
For sequence analysis of LAMP products, to confirm reaction, the lowest band (Fig. 3A) from the LAMP reaction was eluted from a 4% agarose gel using a QIAquick Gel Extraction Kit (Qiagen), and then cloned and transformed using the Topo TA Cloning Kit (Invitrogen, Carlsbad, CA). The plasmid was extracted using QIAprep Spin Miniprep Kit (Qiagen) and sequenced.

Confirmation of loop-mediated isothermal amplification (LAMP) product sequence. (A) Electrophoresis analysis of LAMP products on 4% agarose gel. Lane M, 100 bp DNA marker. The amplified product of red square was eluted for sequence analysis. (B) Result of LAMP product sequence. The arrows were indicated as FIP and BIP primers and extension direction and, the black lines; as region of F1c and B1c primers, the red square; F3 and B3 primers for target region in LasAI gene, respectively.
Comparison analysis of sensitivity of conventional PCR, real-time PCR and LAMP
Because CLas cannot be cultured, purified and quantified genomic DNA of CLas was not available for use as a positive control for real time PCR. Therefore we chose to use DNA extracted from CLas infected grapefruit leaves confirmed by real-time PCR to target tandem-repeats of CLas prophage genes (LasAI and LasAII), ct value was 22.8 in preliminary test. To compare detection sensitivity among conventional PCR, real-time PCR and LAMP, the DNA was serially diluted up to 10−6 and then used as template to evaluate detection limit of conventional PCR, real-time PCR and LAMP.
Conventional PCR
For conventional PCR, the F3/B3 primers listed in Table 1 were chosen. The PCR was performed in a final volume of 20 μl of reaction mixture containing 1 μl template DNA, 2.5 U DNA Taq DNA polymerase (Invitrogen), 1.0 μM of forward and reverse primers, 0.2 mM dNTP, 1 x PCR buffer (20 mM Tris-HCl, pH 8.4 and 50 mM KCl) and 1.5 mM MgCl2. PCR reactions were carried out as follows; pre-denaturation for 3 min at 95°C, 40 cycles of denaturation for 30 sec at 95°C, annealing for 30 sec at 60°C, extension for 30 sec at 72°C, and final extension for 5 min at 72°C. The PCR products were separated by electrophoresis in 1% agarose gel, stained with ethidium bromide and then photographed under UV light using a Gel Doc™ XR+ Gel Documentation System (Bio-rad Labolatories, Inc. Hercules, CA)
Real-time PCR
Additionally, a real-time PCR assay targeting the tandem repeat in LasAI gene of CLas was also conducted using primers and probe listed in Table 1, as previously studied (Zhou et al., 2011). The real-time PCR was performed on a CFX96 Touch™ Real-Time PCR Detection System (Bio-rad Labolatories, Inc. Hercules, CA) and used in a final volume of 20 μl of reaction mixture containing 2 μl template DNA, 1x real-time PCR probe master mix without ROX (GeneSee Scientific, CA, USA), 7 mM MgCl2, 250 nM forward/reverse primers, and 250 nM Taqman probe. The real-time PCR reaction included denaturation for 10 min at 95°C, and was followed by 40 cycles of 15s at 95°C and then 60°C for 30s.
Results
Evaluation of optimal reaction conditions for LAMP
In visual detection assay by adding 2 μl of 1:10 diluted SYBR Green I to mixture, the solution’s color turned green color at 57, 60 and 62°C reaction temperatures in HLB-infected plants as positive reaction while brown color remained at 65°C in HLB-infected plant, healthy plant and negative control as negative reactions (Fig. 2A). When analyzed by agarose gel electrophoresis, ladder-like bands were produced as positive LAMP reactions at 57, 60, and 62°C reaction temperatures in template DNA of HLB-infected plants but not at 65°C in HLB-infected plant, healthy plant and negative control (distilled water) (Fig. 2B).

Loop-mediated isothermal amplification (LAMP) for specific detection of ‘Candidatus Liberibacter asiaticus’ using the primer set from the prophage gene, LasAI in HLB-infected leaves of grapefruit according to reaction temperatures (57, 60, 62 and 65°C). condition test for HLB detection. (A) Visual detection under normal light by adding SYBR Green I dye. (B) Electrophoresis analysis on 1% agarose gel. Lanes 1–4; HLB-infected grapefruit leaves, lanes 5–8; healthy grapefruit leaves, lanes 9–11; distilled water, lane M; 100 bp DNA ladder (NEB New England Biolabs, cat# N3231S).
Sequence analysis of LAMP product
To confirm whether LAMP products from reactions using HLB-infected DNA templates were specifically amplified, the lowest band visible when the LAMP product was run on a 4% agarose gel was cloned, sequenced and compared to the sequence of HLB strain psy62. As a result, the cloned DNA fragment was 182 bp, and the sequences were consistent with those of isolate psy62, within the targeted region of the LasAI gene except for a single mutation found at the 50th base of product (Fig. 3B).
Comparison sensitivity between conventional PCR, real-time PCR and LAMP
Detection sensitivities were compared by evaluation of detection limit by using conventional PCR, real-time PCR and LAMP assay. Specific amplified bands were observed at up to 10−2 dilution in conventional PCR assay. In LAMP, ladder–like bands by 1% agarose gel electrophoresis and green color by adding SYBR green dye were detected at up to 10−3 dilution (Fig. 4). The detection limit of the real-time PCR was at up to 10−3 dilution (Fig. 4D). Detection sensitivity of LAMP was 10 times higher than that of conventional PCR and was same as the sensitivity of real-time PCR.

Sensitivity of loop-mediated isothermal amplification (LAMP), conventional PCR and real-time PCR for detecting HLB. (A) Visual examination of LAMP products by adding SYBR Green I dye. (B) Electrophoresis analysis of LAMP products on 1% agarose gel. (C) Electrophoresis analysis of conventional PCR products on 1% agarose gel. (D) Sensitivity of real-time PCR for detecting HLB using primers (LJ900p, LJ900r)/probe (LJ900p) listed in Table 1. Tube and lane 1–8, and template DNA for real-time PCR; serially diluted genomic DNA (1, 10−1, 10−2, 10−3, 10−4, 10−5, 10−6) and distilled water as negative control, respectively, M; 100 bp DNA ladder.
Discussion
Accurate detection of citrus the HLB-associated bacterium can be challenging because of their low titers and uneven distribution within the infected plants (Li et al., 2009; Tatineni et al., 2008; Teixeira et al., 2008). Current real-time PCR methods target LasAI gene of CLas prophage genes (Morgan et al., 2012). The LasAI gene contained 12 nearly identical tandem repeat (NITRs) 132 bp in length and 4 partial repeats (Puttamuk et al., 2014). When compared with conventional 16S rDNA-based real-time PCR, targeting the repeat sequence reduced the relative detectable threshold by approximately 9 and 3 real-time PCR cycles for LasAI gene (Morgan et al., 2012).
Typical HLB leaf symptoms of citrus trees are very similar to those of micro-element deficiencies, such as zinc, iron and boron, it is difficult to diagnose the disease only the naked eye. Moreover, once infected, it takes more than a year to develop HLB symptoms on trees. Therefore to prevent the spread of HLB, we need a rapid and accurate diagnostic method that can detect the pathogen with low-titer in citrus early in the infection process. Recently, LAMP method is using to detect various plant pathogens including CLas for the advantages of being highly specific, rapid, efficient, and laborsaving. LAMP was developed to detect CLas targeting three copy 16S rDNA and CLIBASIA_ 05175 [GenBank: ACT57606.1] by Okuda et al. (2005) and Rigano et al. (2014), respectively. In this study, we developed LAMP method targeting LasAI gene contained 12 nearly identical tandem repeat for more rapid and sensitive detection of CLas than other methods. Positive results were recorded only for LAMP reactions where HLB-infected samples were used as template both by visual observation of color change within the reaction tubes and by the presence of DNA bands on agarose gels, and sequence analysis of LAMP products were consistent with those of HLB strain psy62, except for a single mutation at the 50th base of the amplicon. It is indicated these LAMP primers used in this study were specific to detect CLas. Though three species, CLas, Clam and CLaf, are associated with HLB, these LAMP primers developed to detect only CLas might be not specifically detect other species, Clam and CLaf.
This new LAMP method was 10 fold more sensitive than conventional PCR in detection of CLas pathogen and similar to that of real-time PCR. The reaction mixtures were reacted at four different reaction temperatures for determination of optimal reaction conditions for LAMP. Positive reactions were achieved in reaction temperature 57, 60 and 62°C but not 65°C. By this result, we recommend 62°C of reaction temperature for this LAMP method.
The LAMP method requires no expensive equipment unlike other PCR methods and its product can be amplified using a single constant temperature. Thus, we expect that this LAMP method shows strong promise as a reliable, rapid, and cost-effective method of detecting the CLas pathogen in citrus and can be applied where rapid diagnosis is needed like such as in the field.
Acknowledgments
This work was carried out with the support of the Cooperative Research Program for Agriculture Science & Technology Development (Project No. PJ01309401), Rural Development Administration, Republic of Korea.