Body
Tomato (Solanum lycopersicum) is one of the most important vegetable crops in the world (Kimura and Sinha, 2008) and is cultivated in different regions of Iran. Tomato production is impacted due to many viral diseases which substantially reduce yield and quality (Golnaraghi et al., 2004).
Orthotospovirus is the only genus of arthropod-vectored viruses in the family Tospoviridae whose members infect different host plant such as tomato, pepper, groundnut, and numerous ornamental plants (10th report ICTV: https://talk.ictvonline.org/taxonomy/). Orthotospovirus’s particles are quasi-spherical, 80-120 nm in diameter and enveloped by a lipid membrane (Chen et al., 2016). The type member of this genus is Tomato spotted wilt virus (TSWV) which has been studied extensively because it has economic impact and broad host range in several important crops especially tomato (Sherwood et al., 2003).
TSWV causes serious damage to a wide range of economically important crops worldwide including fruits, vegetables, ornamental, and weed plants as well (Parrella et al., 2003). TSWV isolates are transmitted by several species of thrips (Thysanoptera, Thripidae) in a persistent and propagative manner, especially the western flowers thrips Frankliniella occidentalis (Jones, 2005; Parrella et al., 2003; Whitfield et al., 2005). Various symptoms including chlorosis, necrosis and ring spots on leaves, fruit, and steams that lead to wilting and death of infected plants are more common in TSWV infection (Tripathi et al., 2015).
The genome of TSWV consists of three single stranded RNA segments denoted as L (8.9 kb), M (4.8 kb), and S (2.9 kb) RNAs which together encode five proteins (Kaye et al., 2011). The L RNA is of negative polarity and encodes a RNA polymerase on the viral complementary strand, which plays an important role in viral replication. In contrast, the other two genomic RNAs use an ambisense coding strategy. The M RNA encodes a nonstructural (NSm) protein in the viral sense and a protein to serve as the precursor for the GN and GC glycoproteins in the viral complementary sense. The NSm protein is involved in cell-to-cell movement of non-enveloped ribonucleocapsid structures. The S RNA encodes in the viral sense, a nonstructural (NSs) protein that functioned as RNA silencing suppressor and in the viral complementary sense, a nucleocapsid protein (NP) that encapsidates viral RNAs (King et al., 2011; Margaria et al., 2015; Takeda et al., 2002).
Tomato spotted wilt virus was first described in 1919 on tomato in Australia (Brittlebank, 1919; Samuel et al., 1930) and since has been reported from many other countries (Lian et al., 2013). Recently, TSWV was detected in Middle-Eastern and Far-Eastern countries in Asia including Japan, South Korea, and China (Zhang et al., 2016).
Tomato spotted wilt virus was reported from Iran based on symptoms and electron microscopy in 1996 (Bananej et al., 1996). There are some reports of Orthotospoviruses from different parts of Iran in tomato, potato, soybean, and ornamental plants based on serological methods and host plants reactions (Ghotbi and Shahraeen, 2012; Golnaraghi et al., 2007; Hajiabadi et al., 2012; Pourrahim et al., 2001).
Limited information is available on the occurrence and sequence characteristics of TSWV in tomato fields from Iran. It is clear that survey for TSWV infection, sequence analysis of the virus and differentiation of viral isolates provide useful information for genetic characterization of the isolates based polymerase gene, NSs, and N genes. Hence, the aim of this study was to investigate the occurrence and genetic diversity of TSWV isolates in tomato fields in Iran, and determine the evolutionary model and genetic relationships of Iranian isolates compared to known TSWV isolates.
Materials and Methods
Virus Source
During spring through fall in 2015 and 2016 around 20 tomato fields were visited to collect the suspected samples with TSWV symptoms. Samples (N=171) were collected based on symptoms such as spots, chlorosis, malformations, necrosis on leaves, and spots on fruits from tomato fields in different regions from West and Northwestern of Iran. A part of samples was stored at −80°C for RNA extraction, and fresh samples were inoculated on different plants for under greenhouse conditions.
Experimental Host range
Host range studies and symptomatology were carried out under greenhouse conditions. The host species that were inoculated included Cucumus sativum (Cucumber), Cucurbita pepo (Squash), Vigna unguiculata (Cowpea), Solanum lycopersicum var Early Urbana and, S. lycopersicum var Superchief (Tomato), Nicotiana benthamiana and, N. tabacum cv. samsum (Tobacco) (Table 1).
Serological Assay for detection of TSWV
Initially, double antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) (Clark and Adams, 1977) was used using polyclonal antibodies (Agdia, USA) against TSWV (diluted 1:1000). An absorbance value at (405 nm) was measured with using an ELISA microplate reader (BioTek ELX 8000, USA). The threshold was set as twice the mean absorbance value of the healthy plant sap.
RNA extraction and RT-PCR
Total RNA was extracted from 100-200 mg of leaves or fruit tissue samples using RNX-plus solution (SinaClon, Iran) according to manufacturer’s instruction and finally suspended in 25 μl sterile distilled water. Optical density (OD) measurements of each extraction were taken to quantify the RNA concentration (ng/μl) and purity (at 260/280 and 260/230 wavelength ratios) using the NanoDropb 2000 (Thermo Scientific, USA). Then, the reverse transcription reaction of 1 μg total RNA was done using Hyperscript master mix (Genall, South Korea) with random hexamer primer in a total volume of 10 μl, according to manufacturer’s instruction.
To test the presence of TSWV, a pair of universal primers (gl3637/gl4435c) corresponded to a conserved region in Orthotospovirus polymerase L gene (Chu et al., 2001) was used for the amplification of an 819 bp fragment. Additionally, two pair primers were designed for RT-PCR detection of TSWV (Table 2), for the N and NSs genes of TSWV to amplify DNA fragments of about 777 bp and 724 bp, respectively. Primer pairs were designed by aligning several sequences of TSWV taken from GenBank (www.ncbi.nlm.nih.gov) using ClustalW alignment in MEGA version 7 (Table 2).
PCR products were amplified under the following conditions: 2 min at 94°C; 35 cycles of 30 s at 94°C, 45 s at 50°C (for universal primers) or 45 s at 52°C (For N and NSs genes primers), and 1 min at 72°C. A final polymerization step at 72°C for 10 min was also applied in three cases.
Cloning
Amplified fragments resulting from the reverse transcription polymerase chain reaction (RT-PCR) were ligated into the pTG19-T vector (Vivantis, Malaysia) using 1 Unit of T4 DNA ligase (Vivantis, Malaysia) according to the manufacturer’s protocol and incubated at 4°C overnight. Ligation mixes were transformed into prepared chemically competent Escherichia coli strain DH5α cells by heat shock method (Chung et al., 1989). The transformed cells were selected on LB plates containing ampicillin (100 mg/ml), IPTG (100 μl of 0.1 M per plate), and X-Gal (20 μl of 50 mg/ml). Plasmids were extracted by Accuprep nanopluse plasmid mini extraction kit (Bioneer, South Korea) according to manufacturer’s instruction. The recombinant plasmids were verified by PCR colony and each colony carrying the cloned cDNA was subjected to single colony isolation. Also, the purified plasmids were digested by BamHI enzyme (Thermo Fisher Scientific, USA) to release the DNA inserted and loaded on 1% agarose gel. Next, the independent clones of each viral isolate were subjected to nucleotide sequencing with M13F/R primers. The sequencing was done by Bioneer Inc. (Seoul, South Korea).
Phylogenetic analysis
The obtained sequences (Nucleotide and amino acid) were analyzed and compared with those available in the GenBank using BLAST program. The sequences were aligned with TSWV isolates around the world (Table 3) by MEGA7 and algorithm of ClustalW and the distance matrix based on Jukes and Cantor’s model was used to estimate nucleotide divergence. The phylogenetic relationships were determined with Neighbor-Joining (NJ) (Saitou and Nei, 1987). The robustness of the inferred evolutionary relationships was assessed by 1000 bootstrap replicates for phylogenetic analysis based on the part of L, N, and NSs genes. All branches with < 50% bootstrap were collapsed. Nucleotide identity and similarity were determined using the MEGA7 software. In addition, Sequence Demarcation Tool version 1.2 (SDT v1.2) (Muhire et al., 2014) was used to generate pairwise nucleotide sequence identity matrix.
Genetic diversity analysis
DnaSP version 6.10.01 (Rozas et al., 2017) was used to provide the confidence intervals of the number of haplotypes (H), haplotype diversity (Hd), number of polymorphic (segregation) sites (S), total number of mutations η (Eta), average number of nucleotide differences (k), average pairwise nucleotide diversity (π), total number of synonymous sites (SS), total number of nonsynonymous sites (NS), and the ratio of non-synonymous nucleotide diversity to synonymous nucleotide diversity (ω) known as ω=dN/dS. There are three types of selection pressure, including negative (purifying), neutral, and positive (diversifying). The gene is under positive, neutral, and negative selection when ω ratio is > 1, = 1 and < 1, respectively (Rozas et al., 2017). To calculate individual codon positions under natural selection was used of single likelihood ancestor counting algorithm (SLAC) which defined in the free and online Datamonkey webserver (http://www.datamonkey.org) within HyPhy software package (Pond and Muse, 2005).
Neutrality, genetic differentiation and gene flow statistical tests
To investigate the neutral selection hypothesis operating by Tajima’s D (Tajima, 1989), Fu and Li’s D* & F* (Fu and Li, 1993) statistical tests and to calculate statistical tests of population differentiation including KS*, KST*, Z*, Snn and FST (Hudson, 2000; Hudson et al., 1992) between phylogroups and geographical populations were done using DnaSP v.6.10.01 for both N and NSs genes. Tajima’s D test static is based on the differences between the number of segregating sites and the average number of nucleotide differences (Tajima, 1989). Fu and Li’s D* test is based on the differences between the number of singletons and the total number of mutation. Fu and Li’s F* test statistic is based on the differences between the number of singletons and the average number of nucleotide differences between pairs of sequences (Fu and Li, 1993). If there is no genetic differentiation (under the null hypothesis), KST* is expected to be near zero, but if it is supported by a small P value (< 0.05) the null hypothesis is rejected (Tsompana et al., 2005). The Z* static is an algorithmic variant of the Z static. Also, if it is too small and supported by significant P value (< 0.05) the null hypothesis is rejected (Hudson et al., 1992). The nearest neighbor of sequences is measured by the Snn test static, whose P value is ranged between 1 (when population is distinctly differentiated) to one-half in the case of panmixia (Hudson, 2000). Finally, the coefficient of FST (genetic differentiation) was used to estimate inter-population diversity and the absolute value of FST ranges between zero to one for indicating undifferentiated to fully differentiated populations (Hudson et al., 1992; Tsompana et al., 2005). Normally, FST > 0.25 suggests a large gene flow (genetic differentiation) within the populations (Gao et al., 2016).
Results
Field observation and host range
During the tomato-growing season, virus symptoms including spots on the fruits and leaves, stem necrosis, chlorosis, leaf deformation and yellow spot discoloration on ripe fruits and subsequent wilting and complete collapse of plants were observed in the inspected fields (Fig. 1) that based on virus descriptions, they seemed to be infected by TSWV. Generally, necrotic spots were observed at young stages of tomato plants on leaves. At the same time that leaf symptoms were developed in the fields, fruit symptoms such as yellow spot were observed in some fields (Fig. 1). It should be noted that some symptoms such as mild and sever mosaic, leaf deformation and shoe string were observed in the fields with common symptoms of TSWV in some samples. A wide range of the observed symptoms in the fields may occur due to mixed infection with other viruses such as Cucumber mosaic virus (CMV) and Tobacco mosaic virus (TMV). Despite the fact that we expected to see the large population of vectors (different species of thrips) in the fields, the vectors observed in some fields as a small population.
The result of mechanical inoculation of plants with sap from field samples showed a range of symptoms at 10-20 days post inoculation (dpi) in different hosts (Table 1). Cucumis sativus and Cucurbita pepo, showed chlorotic lesions after 10 to 15 dpi; whereas systemic symptoms such as leaf deformation, mottling, and necrotic lesions were seen on Vigna unguiculata after 20 dpi (Fig. 2). Necrotic local lesions on Solanum lycopersicum (var Early Urbana and Superchief), and chlorotic spots on Nicotiana benthamiana and N. tabacum cv. samsum were visible 15 to 20 dpi (Fig. 2).
Serological analysis
TSWV was detected by DAS-ELISA in infected leaves and fruits samples from several tomato fields. This infectivity assay provided additional evidence as to the identity of the infecting virus being TSWV.
RT-PCR
The results of RT-PCR using universal primers showed that the expected DNA fragment of about 819 bp in length corresponding to a part of the polymerase gene (L segment) in 46 out of 171 samples. In addition, a DNA fragment about 777 bp encompassing the complete N gene was amplified in 14 out of 46 samples using specific primers. No amplification was obtained from healthy samples that were used as a negative control. Also, by using specific primers corresponding to a part of the NSs gene a DNA fragment of about 724 bp was amplified in 10 out of 14 samples. No DNA fragment was amplified from the healthy control. Optimal amplification conditions included annealing temperature of 50°C, 52°C, and 52°C for amplification of polymerase, N, and NSs genes, respectively. No DNA fragment was amplified from the samples that were negative in DAS-ELISA.
Phylogenetic analysis
Blast analysis of our new nucleotide sequences (Table 3) with those in the GenBank, based on the high levels of sequence similarities, revealed that the new sequences belonged to TSWV. The identities of a part of L polymerase gene were 94-99% between the Iranian and other reported isolates in GenBank whereas the identities among the new TSWV isolates from Iran were 98% to 99% based on nucleotide sequences. Also, alignment of deduced amino acid sequences showed that the identities between the Iranian isolates and other isolates were around 98-99%.
For N gene, the identity of nucleotide and amino acid sequences of the complete N gene for the new Iranian isolates showed 99% and 98% identity, respectively. The Iranian isolates were most closely to six Italian isolates (Table 3), one isolate from USA (AY744478), and two isolates from France (FR692838, FR692839) at 98.3-98.8% nucleotide sequence identity when the new Iranian isolates compared to isolates from elsewhere in the world based on the complete N gene. Italian isolate showed the highest similarity (98.8%) with Iranian isolates (Fig. 3A).
The nucleotide sequence of partial NSs gene of the new Iranian isolates showed 94-99.3% identities to isolates around the world with most closely related to three isolates from France (FR692838, FR692839, FR693033), and two isolates from Italy (DQ431237, DQ398945) at 98.7-99.4% nucleotide sequence identity. The sequences that obtained from a part of NSs gene in this study showed high sequence identity (98.8-100%) among themselves (Fig. 3B). Pairwise nucleotide sequence identity matrix showed the identity result as matrix (Fig. 3B).
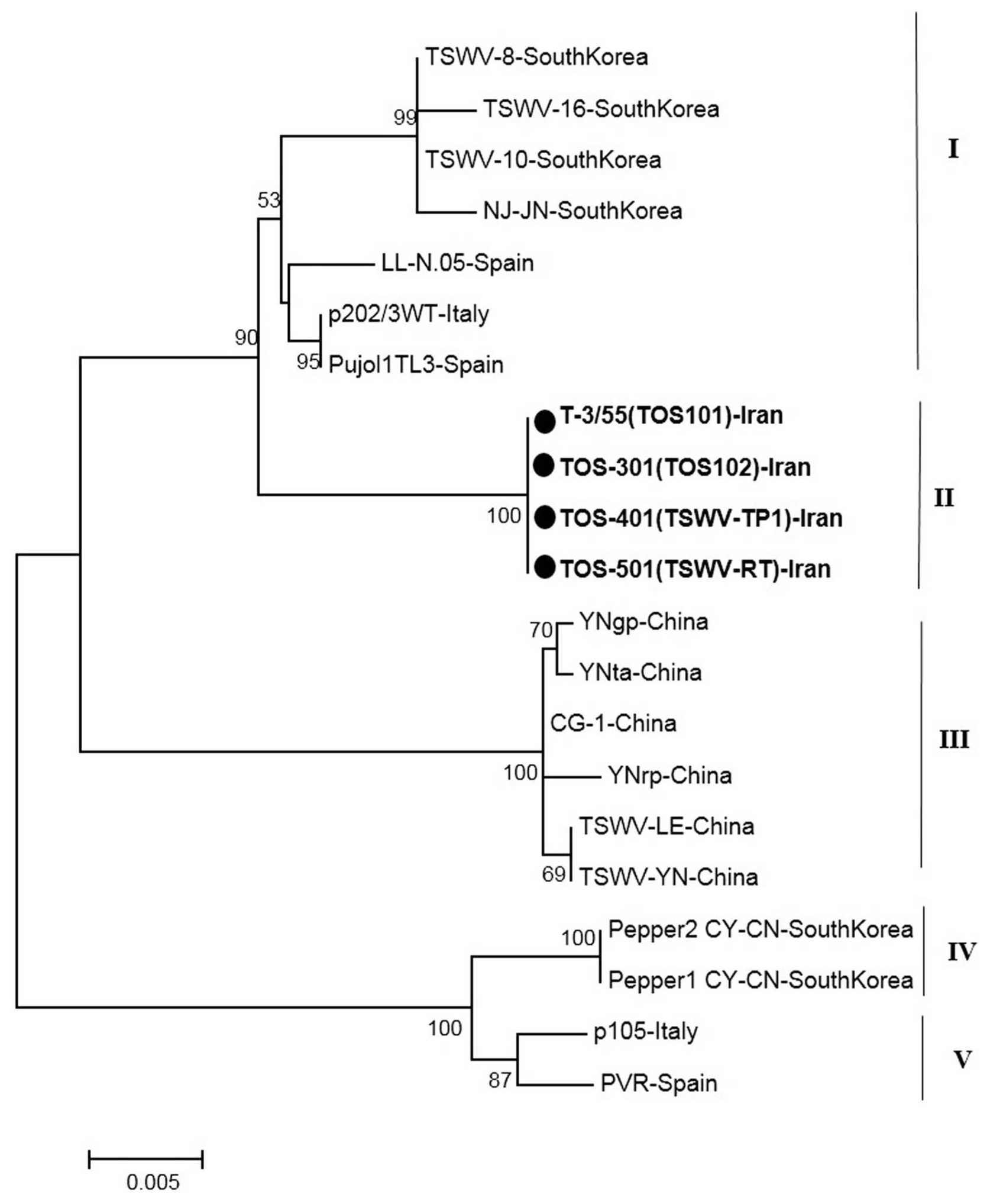
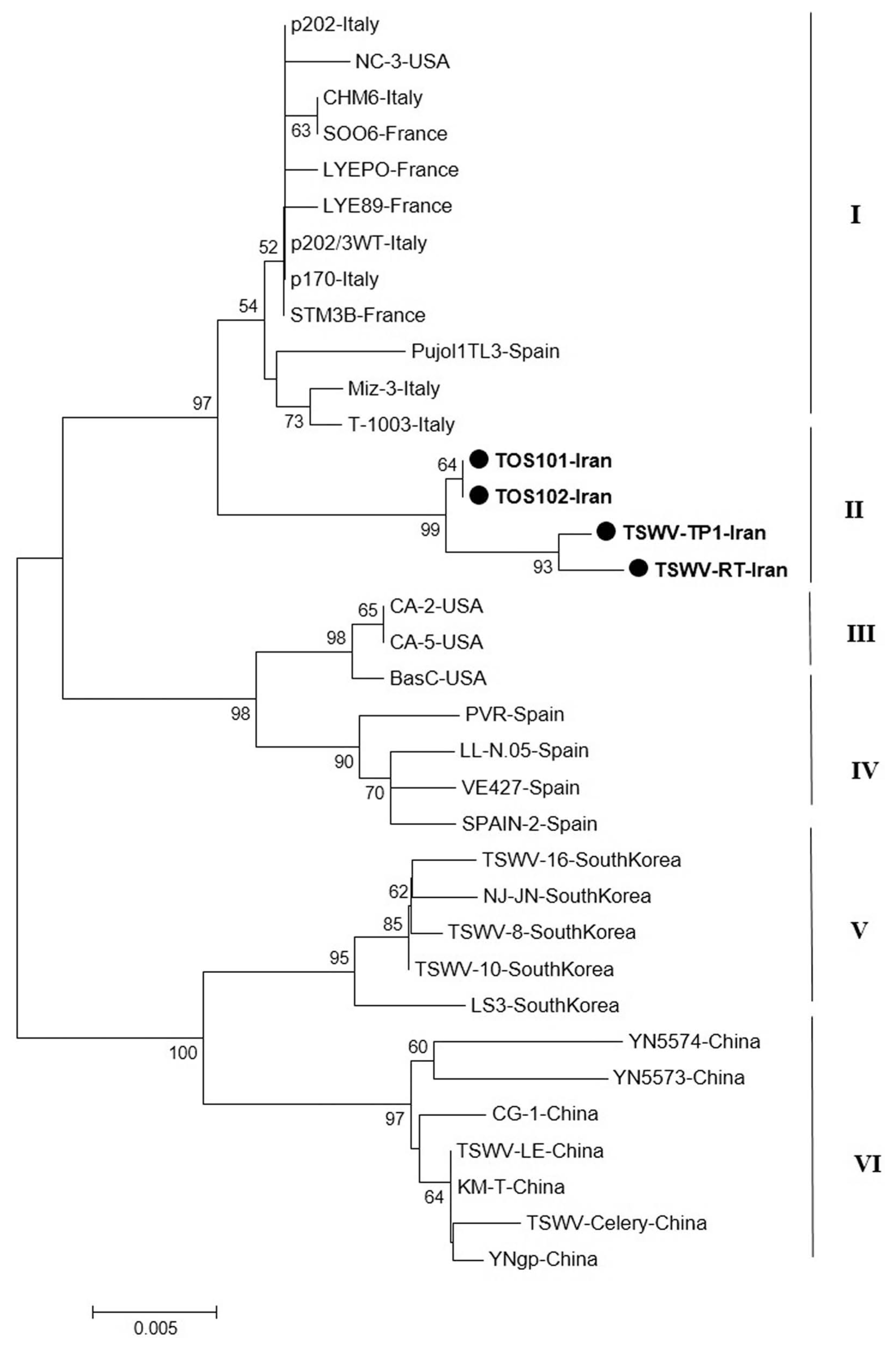
The phylogenetic tree based on a part of L segment consisted of five main clusters (I, II, III, IV and V) that were divided again into several branches (Fig. 4). The cluster I contained the Euro-Asian isolates from South Korea and Spain. The new Iranian sequences were grouped a distinct cluster in Cluster II. All isolates in cluster III originated from China. Other main clusters (IV and V) contained Asian isolates (South Korea), and European isolates (Spain and France) as two distinct clusters (Fig. 4).
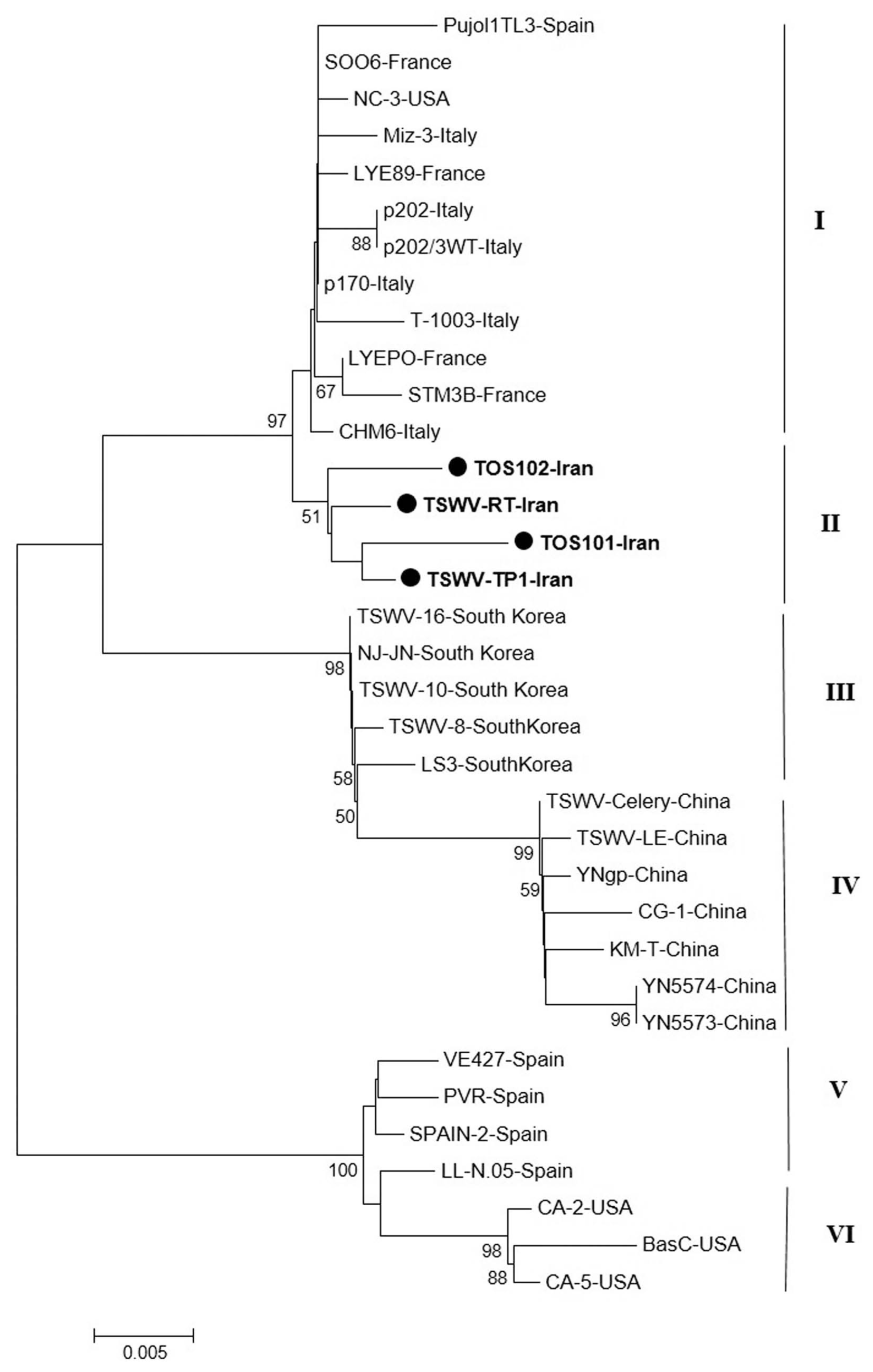
The result of phylogenetic analysis based on the nucleocapsid gene for 31 isolates around the world and four new isolates from Iran with Neighbor-Joining (NJ) method based on the nucleotide sequences produced six independent clusters namely I, II, III, IV, V and VI (Fig. 5). Cluster I include isolates from Italy, Spain, France, and USA; cluster II include new isolates from Iran as a distinct cluster. The main isolates in cluster III were isolates from USA and four Spanish isolates were grouped in cluster IV. South Korean and Chinese isolates were formed two independent clusters V and VI based on the N gene. As noted before, the Iranian isolates formed a single clade distinct from other isolates based on the N gene and were more closely related to isolates from Italy and Spain.
The phylogenetic tree based on nucleotide sequences of the NSs gene of TSWV isolates including four new isolates from Iran produced six main clusters consisting of cluster I with isolates from Europe (Spain, France, and Italy) and one isolate from USA. The Iranian isolates were grouped in independent cluster II that are near to isolates from in cluster I. Cluster III and IV are consisted of all isolates from East-Asian that South Korean isolates were grouped in cluster III and cluster IV is include Chinese isolates. Cluster V include isolates from Spain and USA isolates were grouped cluster VI based on the NSs gene sequences (Fig. 6).
The phylogenetic tree based on the sequences of N and NSs genes of isolates was grouped all isolates in six main groups include cluster I, II, III, IV, V, and VI (Fig. 7) as two other trees based on the N and NSs genes independently. The new Iranian isolates were grouped into a distinct cluster II (Fig. 7). Cluster III include all isolates from USA. The isolates from Spain were grouped into cluster IV (Fig. 7). Like other tree East-Asian isolates were formed two independent clusters that cluster V originated from South Korea and cluster VI were formed Chinese isolates.
Population genetic and polymorphism of TSWV
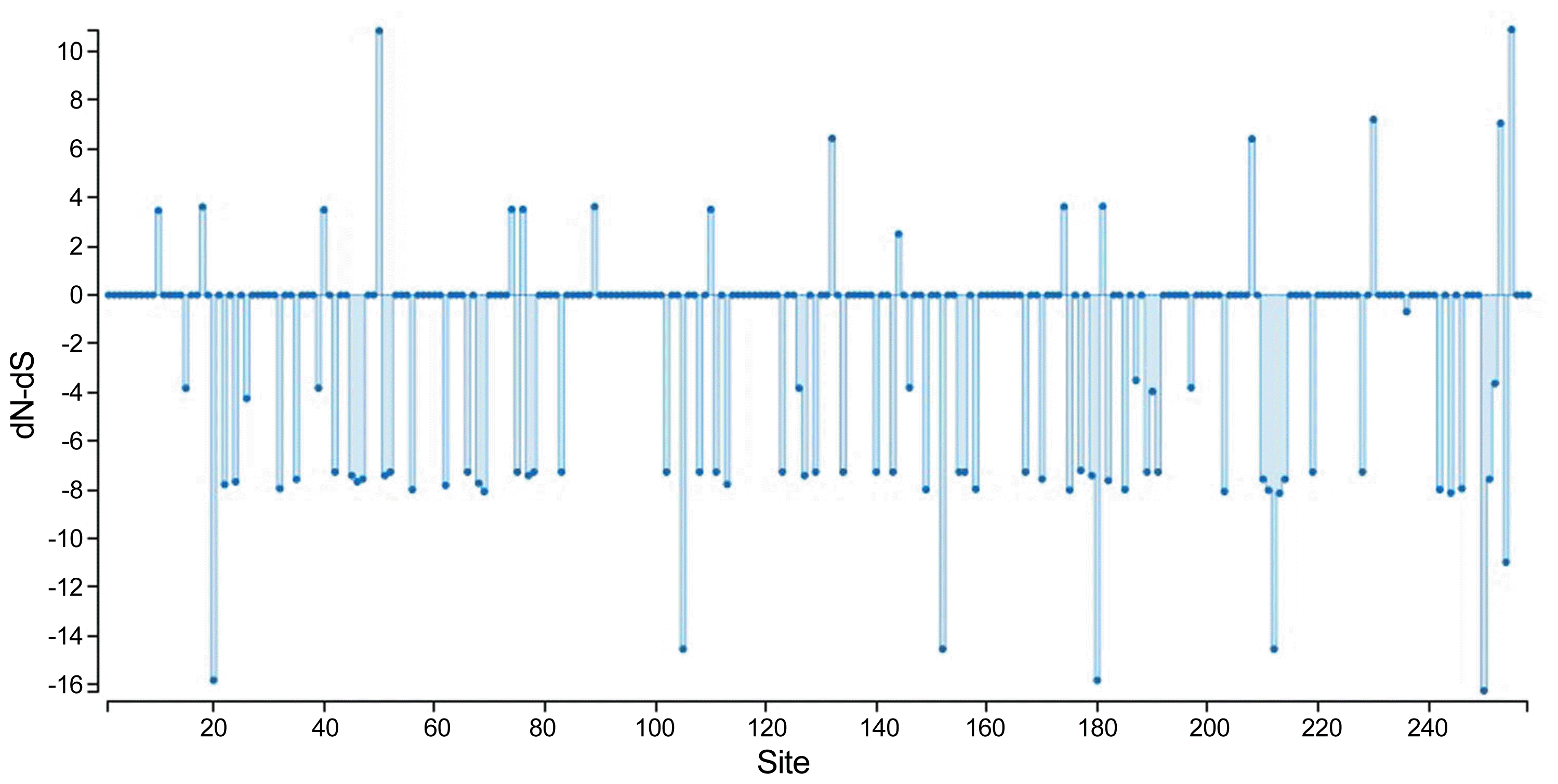
To obtain genetic variety of the TSWV populations based on the N and NSs sequences, several genetic diversity parameters were calculated (Tables 4, 5). The largest average numbers of differences, K (11 and 16 nucleotides) and also the greatest overall nucleotide diversity, π (0.015 and 0.023) between TSWV isolates were calculated for the Spanish and USA populations for N and NSs genes, respectively (Tables 4, 5). Also, the largest number of segregating sites S (28 and 32), and mutations within the segregating sites η (28 and 32) were found in Spanish and USA TSWV populations for N and NSs genes, respectively (Tables 4, 5). Also, the smallest π (0.002 and 0.002) and k (2 and 1) were estimated for French and South Korean populations for N and NSs genes, respectively. Furthermore, the ratio of dN/dS (ω) was < 1 for all populations. The maximum ω ratios were obtained for TSWV from Iran (0.484) and China (0.329) for N and NSs genes, respectively, whereas the minimum ω values were calculated from the France (0 and 0) populations in both N and NSs genes. The results showed that both genes of TSWV populations are under negative selections. The highest (0.484) and lowest (0) ω values were calculated for clades II and III, respectively based on N gene. Also, clade V and clade I were introduced as highest (0.329) and lowest (0.101) ω values in NSs gene, respectively. The level of polymorphism analysis by “Sliding window” options window (50) and step size (25) were high at nucleotides 26-75, 51-100, 201-250, and 526-575 belong to the N gene with the π values of 0.039, 0.038, 0.040, and 0.051, and about NSs gene, were high at nucleotides 26-75, 101-150, and 454-503 with the π values of 0.041, 0.042, and 0.049 respectively (Figs. 8A, 8B, Supplementary). Based on the N gene there were no codons under positive selection by SLAC method in HyPhy software package which implemented in Datamonkey webserver with the p-value threshold (p ≥ 0.1) (Fig. 9, Supplementary). This result showed that the nucleocapsid gene in TSWV is under strong negative evolutionary constraints.
Neutrality tests of TSWV
To explore the molecular variation patterns from segregated sites of TSWV populations based on the N and NSs sequences, several test statics such as Tajima’s D, Fu and Li’s D* and F* (without an out group) were done (Tables 6, 7). Results showed that significantly negative values were obtained only for clade I in the NSs gene and non-significantly positive values were obtained for other clades in both genes (Tables 6, 7). Among geographic populations, non-significantly positive values were found only for Iranian population in both genes in all three test statics and significantly negative value was calculated only for Spanish population in Fu and Li’s D* statistical test in NSs gene (Tables 6, 7).
Gene flow and genetic differentiation of TSWV populations
Gene flow and genetic differentiation analysis of TSWV populations showed that six phylogroups in both genes (N and NSs) with significant KS*, KST*, and Z* are completely distinct. Also, the related Snn values were significantly high (mostly 1.000 and/or near 1.000) (Tables 8, 9). The FST value between phylogroups was > 0.67 and > 0.30 for N and NSs genes, respectively. The highest FST values (0.874 and 0.900) were obtained for South Korea versus France populations for N and NSs genes, respectively, and the lowest FST values (0.027 and 0.052) were found when comparing the France and Italy populations (for N gene) and the USA and Spain populations (for NSs gene), respectively (Tables 8, 9). Among TSWV populations based on the NSs gene, non-significant Z* values were observed between Iran population with USA population, and France population with USA and Italian populations (Table 9). Also, non-significant Z* value was obtained between France population with Italy population based on the N gene (Table 8). Furthermore, among Iranian populations, non-significant Snn value was found between Iran and USA populations and the highest FST value (0.856) was calculated from Iran population with South Korea population in comparison with other Iranian populations in the N gene, and based on the NSs gene, non-significant Snn values were obtained between Iran population with USA and French populations and the highest FST value (0.808) between Iranian populations was found when comparing the Iran and China populations. In both genes FST values were higher than 0.25 (Tables 8, 9). Between phylogroups, the maximum FST values were evaluated for clade I versus clade III (0.886) and clade III versus clade VII (0.909) for N and NSs genes, respectively, and the minimum FST values were obtained between clade III and clade V (0.666) (for N gene) and clade I and clade II (0.300) (for NSs gene) (Tables 8, 9).
Discussion
Viral diseases are one of the serious problems that reduce quality and quantity of tomato crops in Iran. In the present research, we observed a variety of viral symptoms including mild and sever mosaic, necrotic spots on the leaves, yellow spots on the fruits, malformations, and chlorosis in the different tomato fields of West and Northwest in Iran incurring significant losses. The study confirmed the presence of TSWV in West and Northwest of Iran, even though there are some reports of TSWV from Iran based on symptoms and serological tests in tomato, soybean, potato, tobacco, peanut, and ornamental plants (Golnaraghi et al., 2001a, 2001b; Pourrahim et al., 2001). However, the genetic variation of TSWV isolates from tomato has not been studied. On the other hand, the symptoms of TSWV on infected tomato leaves and fruits are similar to TYRV that was reported from Iran. It is possible that similarity of symptoms to TYRV is due to the cross reactions of different species in the same serogroup (Harrison, 2002; Pang et al., 1994) and therefore serological assays may not detect TSWV properly. In this study we used a more sensitive method and sequences different genomic region of TSWV to confirm TSWV infection and also investigate the genetic variation of TSWV isolates from tomato plants.
As noted before, virus symptoms were observed in most tomato fields that were visited in different regions; however, necrotic spots on the leaves, common mosaic and yellowing were most common symptoms that observed in early summer; whereas yellow spot and ring spot symptoms were observed in middle of summer and autumn. Yellow spots on the fruit symptoms are the typical symptoms caused by TSWV. Different symptoms may show the presence of other viruses and/or mix infections by common viruses. There are other viruses that infected tomato including TSWV, CMV, Tomato mosaic virus (ToMV), TMV, Potato virus X (PVX), Tomato ringspot virus (ToRV), and Beet curly top virus (BCTV) that have been reported in the most regions of tomato fields (Escriu et al., 2003; Hanssen and Lapidot, 2012).
In tomato plants showing a range of symptoms, mixed infection of CMV and ToMV was reported. Notably, for the geographical region that we collected tomato samples, a high rate of CMV infection in the cucurbits and tomato crops has reported (Sokhandan et al., 2008; Valizadeh et al., 2011). Therefore, identifying TSWV based on the symptom is complex and there is a need to molecular methods for detection of TSWV in tomato plants.
The experimental host range assay provided further evidence for the presence of TSWV in the examined isolates because the inoculated plants developed symptoms resembling those of TSWV infected plants and showed positive reactions in the ELISA. However, considering the serological cross reaction between TSWV and other close viral species (Harrison, 2002; Pang et al., 1994;) we still need to confirm the presence of TSWV by a more sensitive and accurate method such as RT-PCR.
RT-PCR using specific primers to different regions of TSWV genome followed by sequencing was the most definitive approach for the identification of TSWV. The degenerate primers designed based on the consensus nucleotide sequences of RdRp initially provided the identification to the genus level (Tsompana et al., 2005). In contrast, the sequence of N genes form Orthotospoviruses are more diverse, thus it can be the key gene target for classification of a Orthotospovirus (Margaria et al., 2015) and it could be reliable to differentiate the Orthotospovirus members because the consensus nucleotide sequences of N genes in Orthotospovirus. The samples that not yield any amplification in RT-PCR raised the possibility that other viruses could be present in those plants. Whether negative samples in this study infected by other viruses need to be analyzed further. Further testing showed the presence of TMV and CMV in the remaining samples (data not shown). Presence of other viruses such as CMV in tomato fields is not unusual in the studied area (Bashir et al., 2006; Sokhandan et al., 2008).
Phylogenetic analysis showed that the new Iranian isolates were grouped into a distinct cluster. The isolates from Italy and France were placed close to the Iranian isolates. Such a phylogenetic relation between the isolates may be related to the virus transmission. In addition of the polymerase gene, some studies have focused on the diversity of N and NSs genes for strain identification and genetic analysis (Chiemsombat et al., 2008; Nischwitz et al., 2007; Pappu et al., 2009; Zheng et al., 2008).
Sequence analysis for N and NSs genes revealed that the Iranian isolates were grouped with several isolates from Europe. The Iranian isolates were differentiated from European isolates by high bootstrap, but separated from South Korean, Japanese, and American isolates. It is possible that origin of Iranian TSWV isolates is different from other Asian countries and they could have been introduced from Europe. As a result, TSWV isolates were not grouped according to geographical origin, as previously reported by (Zindovic et al., 2014), whereas most Asian isolates (South Korea, Japan, and China) were grouped in a distinct cluster. Global trade in plant products might be the main contributing factor for TSWV spread throughout the world.
In addition, the high nucleotide identity and similarities among the Iranian isolates based on the L, N, and NSs genes showed that minor evolutionary differences exist amongst the isolates. However, the minor isolate differences are evidence of some mutations taking place in different genomic regions. Population genetics studies should be conducted to find the systematic and random forces of evolution factors on the new isolates.
It is clear that mutation has a significant effect in shaping the population structure of TSWV (Kaye et al., 2011). Also, survey of the variability in the genetic structure of plant virus populations is crucial for a better understanding of virus evolution and interaction between plant and virus (Tsompana et al., 2005). Hence, detection and sequencing of TSWV isolates have an important implication for crop disease management.
Because TSWV has numerous opportunities to exchange genetic information with other strain or viruses (Kaye et al., 2011), the complete genome sequences of TSWV isolates with molecular diversity will provide fundamental information to elucidate molecular interactions between host plants, TSWV, and geographic regions. Furthermore, this information can be applied practically in molecular breeding to develop TSWV-resistant vegetables and crops.
In this research, genetic diversity and molecular evolutionary were examined to identify haplotypes and infer genetic relationships between populations of the TSWV based on the N and NSs genes. Comparison of nucleotide diversity (π) showed that the lowest nucleotide diversity (0.002) was found for clade III in comparison with other clades and among geographical populations was obtained for French TSWV population (0.002) based on the N gene. Total of nucleotide sequence diversity was 0.064. One of the most important reasons that may have caused the nucleotide diversity of this phylogroup and France population to be less than other phylogroups is due to strong negative selection (ω=0) (Yu et al., 2010). The largest dN/dS ratios were found for Iran population (0.484) and clade II (0.484) in the N gene which isolated from different varieties of Soalnum lycopersicum, and based on the NSs gene, the maximum dN/dS ratios were obtained for China population (0.329) and clade V (0.0329) from different hosts (Table 3). The obtained ω ratios were less than 1 for all populations, suggesting that the negative selection has been occurred for all the TSWV populations. Some studies on viral N genes in other RNA viruses have revealed that this gene can be conserved under negative selection (Gao et al., 2016).
Haplotype diversity (Hd) measures can range from 0, meaning no diversity, to 1.000, which indicates high levels of haplotype diversity (Nei and Tajima, 1981). Our data showed that Hd was 0.983 to 0.990, indicating very high levels of diversity for each locus. These levels were similar to those indicated for global populations, which were 0.857 to 1 (Tsompana et al., 2005). Most studies of Orthotospovirus spp. have focused on the genetic diversity of the N gene for strain identification or the NSs gene (Chen et al., 2006; Pappu et al., 2006). In addition, these studies should include noncoding regions of the TSWV genome to provide enhanced statistical analysis of the data (Rosenberg and Nordborg, 2002).
Nucleotide diversity analysis by “Sliding window” option with 50 and 25 as the window and step size, respectively, revealed high polymorphism at middle regions and 5′ parts of both N and NSs genes (Figs. 8A, 8B, Supplementary). This information can help to avoid designing specific primers for these regions of viral genome for virus detection.
Non-significantly negative and positive values in all clades were calculated by neutrality test statics (Tajima’s D, Fu and Li’s D* and F*) in both genes except significantly negative values for clade I in the NSs gene. Between geographic populations, Fu and Li’s D* test static was only significantly negative for Spanish population in the NSs gene which shows low-frequency polymorphism and only Iranian population was non-significantly positive in both N and NSs genes (Tables 4, 5). In geographical regions, the maximum FST values between South Korea and France populations based on the N (FST = 0.874) and NSs (FST = 0.900) genes could be due to long distances between these countries. The results obtained from phylogenetic analysis and genetic differentiation were in line with each together. For instance, the minimum FST value was obtained between USA/Spain populations (FST = 0.052) based on the NSs gene, and France/Italy populations (FST = 0.027) based on the N gene which they clustered together in a sub-clade in phylogenetic tree (Figs. 5, 6).
Using SLAC algorithm which implemented in Datamonkey server within HyPhy software package, negative selection pressure was observed at position 258 codon and it seems that negative pressure is one of the most important reasons for survival and evolution of the TSWV. In this study and several studies the N gene was under negative pressure (Timmerman-Vaughan et al., 2014).
In summary, the first detailed molecular characterization followed by genetic diversity and population structure studies of TSWV from tomato in Iran has shed light on the nature of TSWV populations in tomato and provides a basis for further studies on TSWV in other crops and toward developing a sound disease management program.





 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Print
Print