Complete Genome Sequences and Evolutionary Analysis of Cucurbit aphid-borne yellows virus Isolates from Melon in Korea
Article information
Abstract
Complete genome sequences of 22 isolates of Cucurbit aphid-borne yellows virus (CABYV), collected from melon plants showing yellowing symptom in Korea during the years 2013–2014, were determined and compared with previously reported CABYV genome sequences. The complete genomes were found to be 5,680–5,684 nucleotides in length and to encode six open reading frames (ORFs) that are separated into two regions by a non-coding internal region (IR) of 199 nucleotides. Their genomic organization is typical of the genus Polerovirus. Based on phylogenetic analyses of complete nucleotide (nt) sequences, CABYV isolates were divided into four groups: Asian, Mediterranean, Taiwanese, and R groups. The Korean CABYV isolates clustered with the Asian group with > 94% nt sequence identity. In contrast, the Korean CABYV isolates shared 87–89% sequence identities with the Mediterranean group, 88% with the Taiwanese group, 81–84% with the CABYV-R group, and 72% with another polerovirus, M.. Recombination analyses identified 24 recombination events (12 different recombination types) in the analyzed CABYV population. In the Korean CABYV isolates, four recombination types were detected from eight isolates. Two recombination types were detected in the IR and P3–P5 regions, respectively, which have been reported as hotspots for recombination of CABYV. This result suggests that recombination is an important evolutionary force in the genetic diversification of CABYV populations.
Body
Cucurbit aphid-borne yellows virus (CABYV) is an important pathogen that causes yellowing symptoms in cucurbit crops worldwide. CABYV is a member of the genus Polerovirus in the family Luteoviridae (D’Arcy et al., 2005; Mayo and d’Arcy, 1999). It is transmitted by aphids, mostly Aphis gossypii and Myzus persicae, with the infection rate of 48–70% in a persistent manner (Kassem et al., 2013; Lecoq et al., 1992). Typical symptoms of CABYV include yellowing and thickening of lower and older leaves. CABYV has been shown to reduce yield by up to 50% in melons (Lecoq et al., 1992). CABYV was first described in melon and cucumber plants in 1992 in France and has since been detected in numerous cucurbit crops in many other countries countries (Abou-Jawdah et al., 1997; Al Saleh et al., 2015; Bananej et al., 2006; Juárez et al., 2004, 2005; Lecoq et al., 1992; Lemaire et al., 1993; Mnari Hattab et al., 2005; Omar and Bagdady, 2012; Orfanidou et al., 2014; Svoboda et al., 2011; Tomassoli and Meneghini, 2007; Xiang et al., 2008a; Yardımcı and Özgönen, 2007).
CABYV has a single-stranded positive sense RNA genome of approximately 5.7 kb nucleotides (nt) comprising six open reading frames (ORFs) that are separated into two regions by a non-coding internal region (IR) of about 200 nt (D’Arcy et al., 2005). The 5′-proximal ORFs (ORF 0, 1, and 2) are translated from genomic RNA and yield the proteins P0, P1, and the ribosomal frameshift protein P1–P2. The 3′-proximal ORFs (ORF 3, 4, and 5) are translated from subgenomic RNA and yield the proteins P3, P4, and readthrough protein P3–P5. P0 protein is a suppressor of post-transcriptional gene silencing (PTGS) (Pfeffer et al., 2002). P1 has regions of amino acid sequence similarity with serine proteases and genome-linked viral proteins (VPgs) of other poleroviruses, and P1–P2 has amino acid motifs typical of RNA-dependent RNA polymerases (RdRP) (Guilley et al., 1994; Mayo and Miller, 1999). P3 is a coat protein (CP); P4 is a movement protein (MP); and P3–P5 is involved in transmission by aphids (Mayo and Miller, 1999).
To date, complete genome sequences of 11 CABYV isolates from France, Spain, China, Japan, and Taiwan have been reported (Kassem et al., 2013; Knierim et al., 2013; Lecoq et al., 1992; Xiang et al., 2008b), as well as partial sequences of ~100 CABYV isolates from several other countries. Based on phylogenetic analysis, CABYV isolates have been divided into two subgroups: the Asian and Mediterranean groups (Shang et al., 2009).
In Korea, CABYV was first detected by next-generation sequencing (NGS) in melons showing yellowing symptoms in 2014, and it was confirmed that the leaf yellowing symptom of melon is not merely a physiological disorder but a viral disease caused by CABYV (Lee et al., 2015).
In this study, we determined the complete genome sequences of 22 CABYV isolates collected from melons showing yellowing symptoms in melon-producing areas during 2013–2014. We analyzed the molecular characteristics and genetic structure of Korean isolates of CABYV in comparison with those of previously reported isolates using a range of methods to understand the evolutionary relationships among isolates.
Materials and Methods
Survey and virus isolates
A survey of CABYV infecting melon was carried out in seven melon-producing areas of Korea during the years 2013–2014 (Fig. 1). We collected 308 samples of melon (Cucumis melo L.) leaves that showed yellowing and mosaic symptoms (Table 1). Samples were maintained at −70°C until analysis of CABYV by reverse transcription-polymerase chain reaction (RT-PCR).
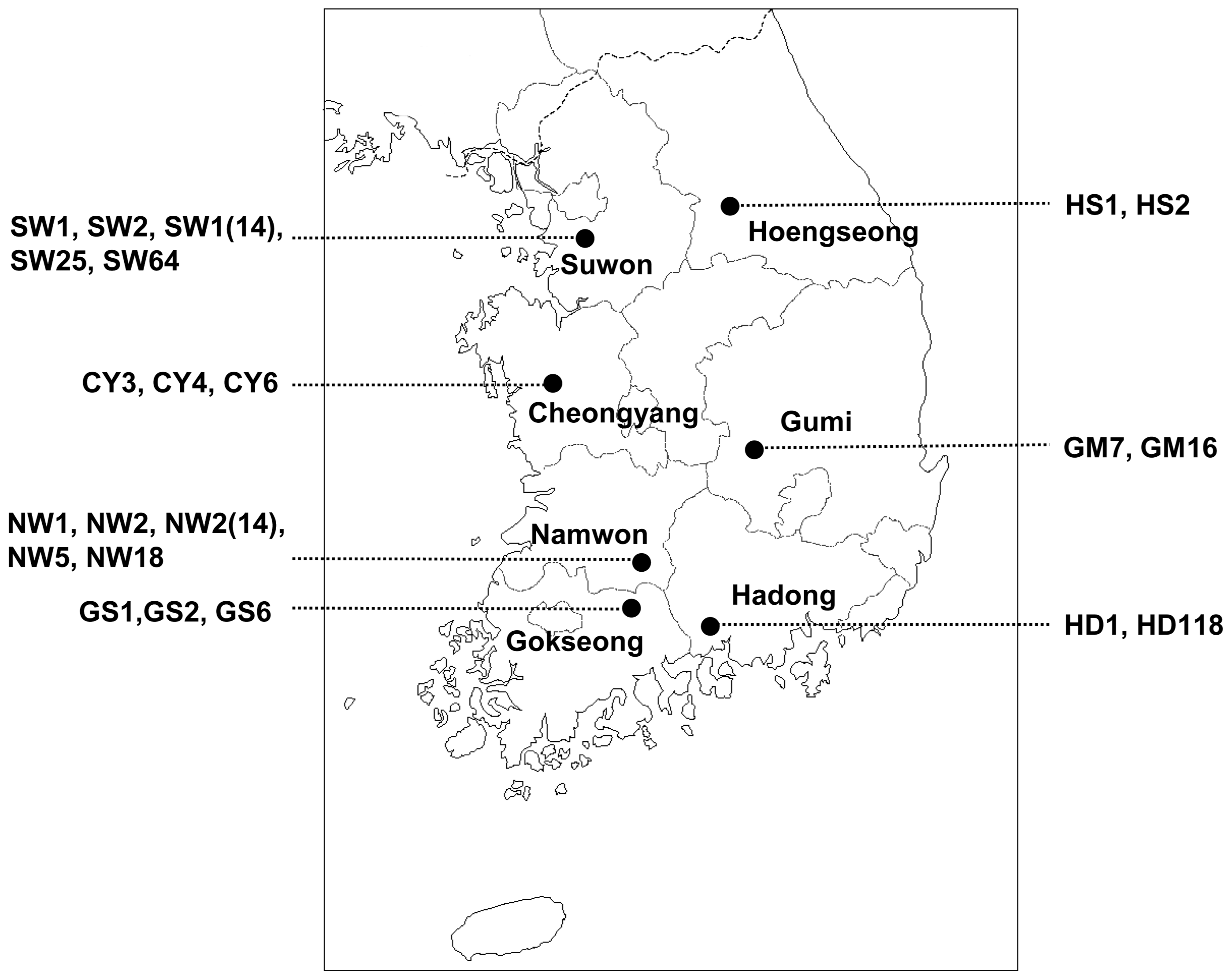
Geographical locations in Korea at which the CABYV isolates were collected.

Survey and detection of CABYV from melon in seven areas in Korea
Of CABYV-positive samples, the full-length genome sequences of the following 22 CABYV isolates were determined: 5 isolates (SW1, SW2, SW1(14), SW25, and SW64) selected from Suwon in 2014, 3 isolates (CY3, CY6, and CY4) from Cheongyang in 2013 and 2014, 5 isolates (NW2, NW5, NW18, NW1, and NW2(14)) from Namwon in 2013 and 2014, 3 isolates (GS1, GS2, and GS6) from Gokseong in 2013 and 2014, 2 isolates (GM7 and GM16) from Gumi in 2013, 2 isolates (HD1 and HD2) from Hadong in 2014, and 2 isolates (HS1 and HS2) from Hoengseong in 2014 (Table 1 and Fig. 1).
RT-PCR, cloning and sequencing
Total RNA was extracted from infected leaf samples using an Easy-spinTM Total RNA Extraction Kit (Intron, Korea) according to the manufacturer’s instructions. RT-PCR was carried out as either one-step RT-PCR (Genetbio, Korea) for CABYV detection, and two-step RT-PCR including RT using AMV reverse transcriptase (Promega, USA) and PCR using high-fidelity LA Taq polymerase (Takara, Japan) for full-length genome sequencing. Pairs of specific primers for the detection and full-length genome sequencing of CABYV were designed based on previously reported CABYV nucleotide sequences and contig sequences determined by NGS techniques (Lee et al., 2015) (Table 2). cDNA clones containing the 5′ end of the genomes were produced using a sense primer (5′-ACAAAAGATACGAGCGGGTGA TGC-3′) complementary to the conserved 24 nt at the 5′ terminus and an antisense primer (5′-GCGAGGAAAAATCGCGCAAC-3′) complementary to nt 352-333 in the CABYV genome. In addition, cDNA clones containing the 3′ end of the genomes were produced using a sense primer (5′-ATGGATARYAGGAAGAAATGGGGA-3′) complementary to nt 5,314-5,337 and an antisense primer (5′-ACACCGAAACGCCAGGGGG-3′) complementary to the conserved 19 nt at the 3′ terminus of the CABYV genome. All the PCR products were overlapped at least 200 bp to ensure that they were amplified from the same genome. Each PCR fragment was purified using a MEGA Quick-spin™ Kit (Intron, Korea) and cloned into the pGEM-T easy vector (Promega, USA) according to the manufacturer’s instructions, followed by transformation into Escherichia coli DH5α. The clones of each fragment were completely sequenced by a commercial company (Genotech, Korea). The resultant sequences were assembled using DNA Star v. 5.02 (Lasergene, USA) and have been submitted to GenBank database under the accession numbers listed in Table 3.
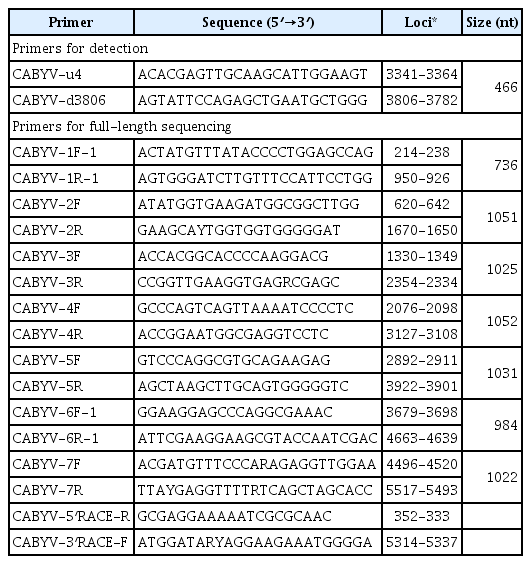
Primer pairs used for detection and full-length sequencing of the CABYV genome
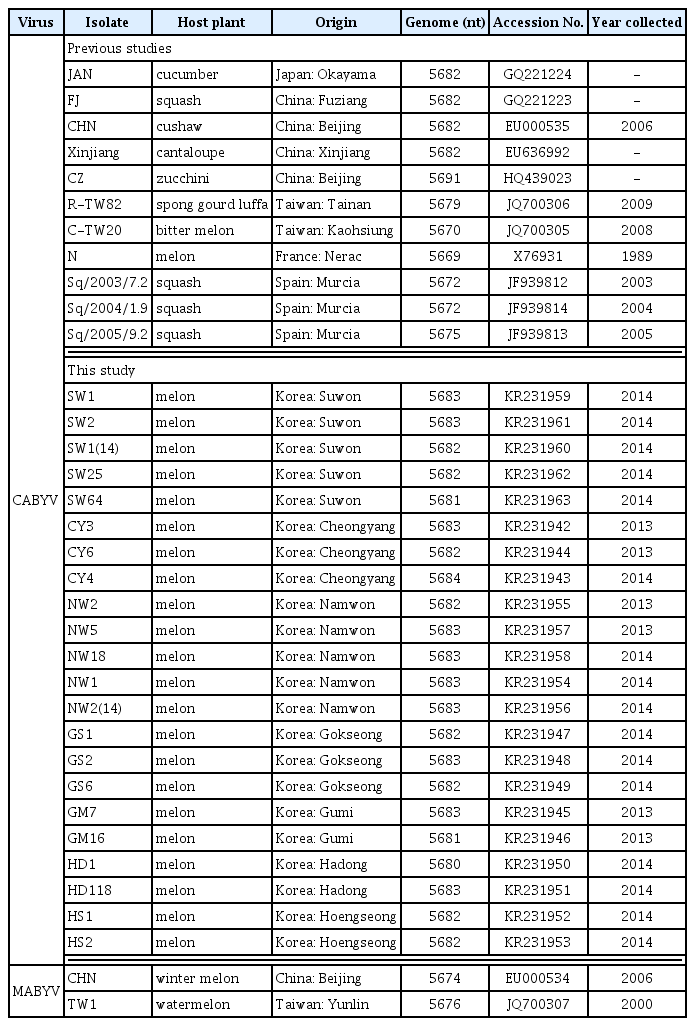
Database of the complete nucleotide sequences of CABYV genomes
Sequence and phylogenetic analyses
The complete nt sequences and the deduced amino acid sequences were aligned using the ClustalX2 program and Geneious methods in Geneious Pro 8 and compared with those of previously reported isolates; i.e., the JAN (Japan), CHN, FJ, Xinjiang, and CZ (China, Xiang et al., 2008b), R-TW82 and C-TW20 (Taiwan, Knierim et al., 2013), N (France, Lecoq et al., 1992), Sq/2003/7.2, Sq/2004/1.9, and Sq/2005/9.2 (Spain, Kassem et al., 2013). The MABYV isolates, CHN and TW1, were used as outgroups (Table 2). Nucleotide and deduced amino acid sequence similarities were analyzed using AlignX implemented in the Vector NTI Suite (Invitrogen, Carlsbad, CA). Pairwise genetic distances and pairwise synonymous (dS) and nonsynonymous (dN) substitutions were analyzed by Kimura’s two-parameter method (Kimura, 1980) and the Pamilo-Bianchi-Li method (Li, 1993; Pamilo and Bianchi, 1993), respectively, using the MEGA6 program (Tamura et al., 2013). The phylogenetic relationships of the CABYV sequences were analyzed by the maximum likelihood (ML) method in MEGA 6. In ML analyses, the phylogenetic trees were constructed using best fit nucleotide substitution models (GTR+G+I for full-length genome and K2+G for IR region and 3′ UTR) and best fit amino acid substitution models (JTT+G for all protein regions). Bootstrap values were calculated using 1,000 random replication. All positions containing gaps and missing data were eliminated. Geneious Pro 8 software was used to calculate the percentage nucleotide and amino acid identities.
Recombination analyses
Recombination events on the full-length sequences of 33 CABYV isolates and 2 MABYV isolates were analyzed using RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan, and 3Seq methods implemented in the RDP4 software (Recombination Detection Program, ver. 4) with default settings and a Bonferroni corrected P-value cut-off of 0.01. To reduce the possibility of false detection of recombination, only recombination events supported by at least three methods were selected. To further investigate the putative recombination signals, phylogenetic network analysis was performed using SplitsTree v. 4.1 program (Huson and Bryant, 2006).
Results
Genome characterization of Korean CABYV isolates
We collected melon leaf specimens that showed yellowing and mosaic symptoms from seven melon-producing areas of Korea during the years 2013–2014. These leaves were analyzed for CABYV using RT-PCR. Of the 308 leaf samples collected, 245 (80%) were positive for CABYV (Table 1). Of the CABYV-positive samples, we selected 22 CABYV isolates based on geographic location, and determined their full-length genome sequences (Table 3). The representative symptoms included yellowing, local chlorosis, mosaic patterns on infected leaves, and informal net formation on fruits (Fig. 2). The complete genomes of Korean CABYV isolates ranged from 5,680 to 5,684 nt, and encoded six open reading frames (ORFs) that were separated into two regions by a non-coding internal region (IR) of 199 nt. Their genomic organization is typical of members of the genus Polerovirus. The 5′ and 3′ non-coding regions (NCR) are 20 and 164–167 nt in length, respectively. The 5′-proximal ORFs (ORF 0, 1, and 2) encode P0, P1, and the ribosomal frameshift protein P1–P2 with sizes of 239 aa, 631 aa, and 1,056 aa, respectively. The 3′-proximal ORFs (ORF 3, 4, and 5) encode P3 (CP), P4 (MP), and readthrough protein P3–P5 with sizes of 199 aa, 191 aa, and 667–668 aa, respectively (Fig. 3A). The genome organization of these Korean isolates is similar to those of other Asian group CABYVs, including CABYV-JAN and CABYV-CHN.

Symptoms induced by CABYV on naturally infected melon plants in Korea. Yellowing (A), leaf mosaic (B), and informal net on melon fruits (C)
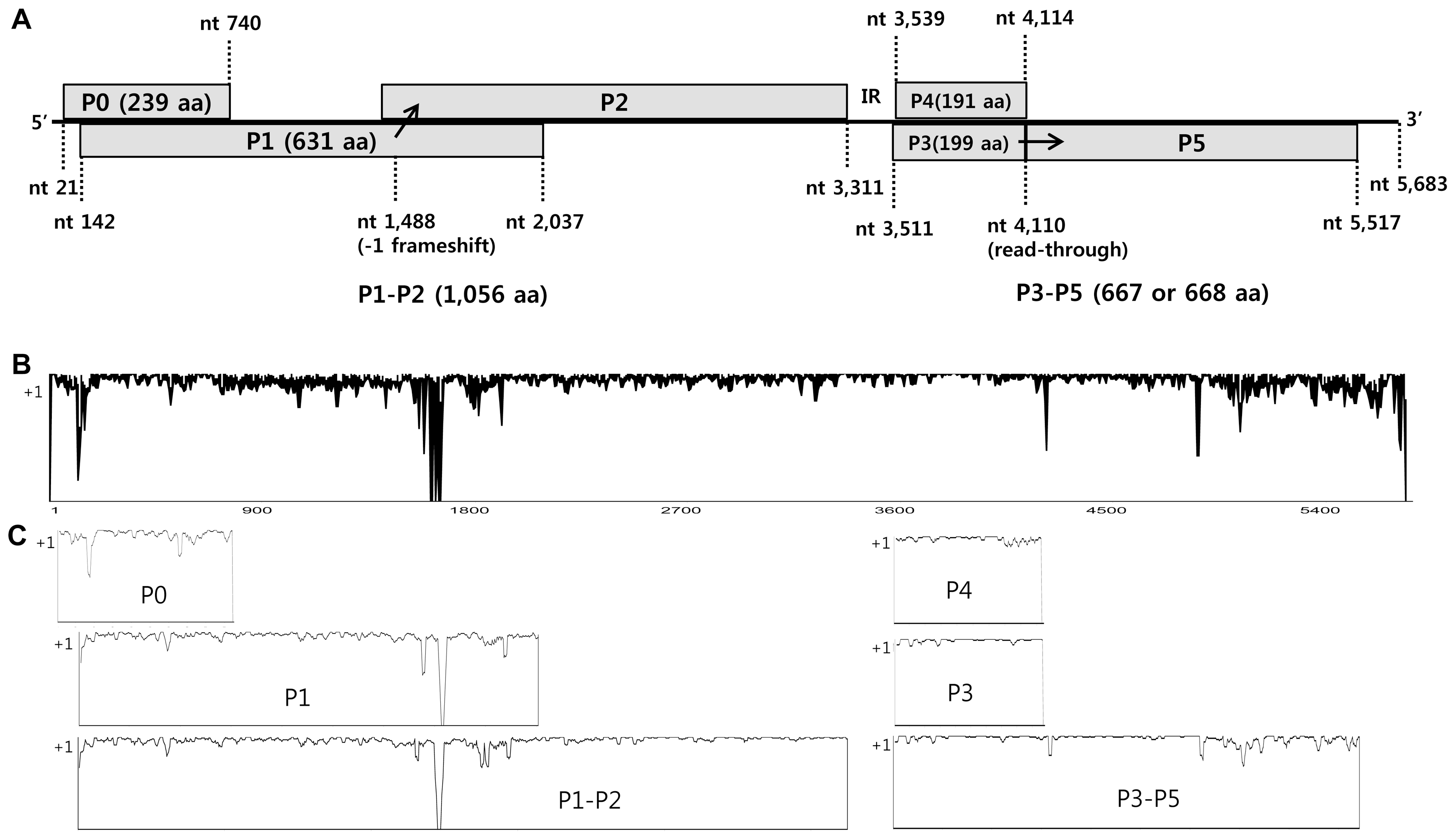
Genome organization of Korean CABYV isolates (A). The six proteins are separated by IR into two regions: 5′ proximal and 3′ proximal proteins. A ribosomal frame shift (−1) in the P1–P2 protein is indicated at nt 1,488 and readthrough of the P3–P5 protein occurs at nt 4,110. Nucleotide (B) and deduced amino acid (C) sequence similarities in the CABYV population. Full-length sequences of 33 CABYV isolates were aligned by ClustalX2 and analyzed using AlignX by setting window site to estimate similarities. ‘+1’ on y-axis means that sequences are perfectly conserved.
Genetic diversity in genome region of CABYV population
The molecular variability of 33 isolates of CABYV population, including 22 Korean CABYV isolates and 11 previously reported CABYV isolates, was compared using both complete nucleotide and deduced amino acid sequences. Widespread nucleotide variations were detected throughout the genomes of CABYV. Especially, more significant variations were observed in 3′-UTR and P0, P1–P2 and P3–P5 regions while P3 (CP) and P4 (MP) region were relatively conserved (Figs. 3B and 3C).
Nucleotide diversity for different genomic regions of the CABYV population was estimated by Kimura’s two-parameter method. Low nucleotide diversity values were observed in CP and MP regions and other regions showed the relatively high diversity values (Table 4). Also, pairwise genetic differences at nonsynonymous (dN) and synonymous (dS) nucleotide position were estimated using the Pamilo-Bianchi-Li method. The ratio between nucleotide diversity values in nonsynonymous and synonymous positions (dN/dS) provides an estimation of the degree and direction of the selective constraints acting on the coding regions of CABYV. On the whole, the values of the dN/dS ratio for all the coding regions except P0 were under 1, indicating that these genes are under negative or purifying selection. While, the ratios of dN/dS for P0 gene was greater than 1, considered as evidence for positive selection.

Nucleotide diversity for different genomic regions of the CABYV population
Analysis of phylogenetic relationships
The complete nucleotide and deduced amino acid sequences of 22 Korean CABYV isolates were compared to those of 11 previously reported CABYV isolates. Two MABYV isolates were included as outgroup isolates in the phylogenetic analyses (Table 3). Full-length genome sequence-based phylogenetic analyses revealed that the Korean CABYV isolates clustered with the Asian group, including Japanese and Chinese isolates (Fig. 4).
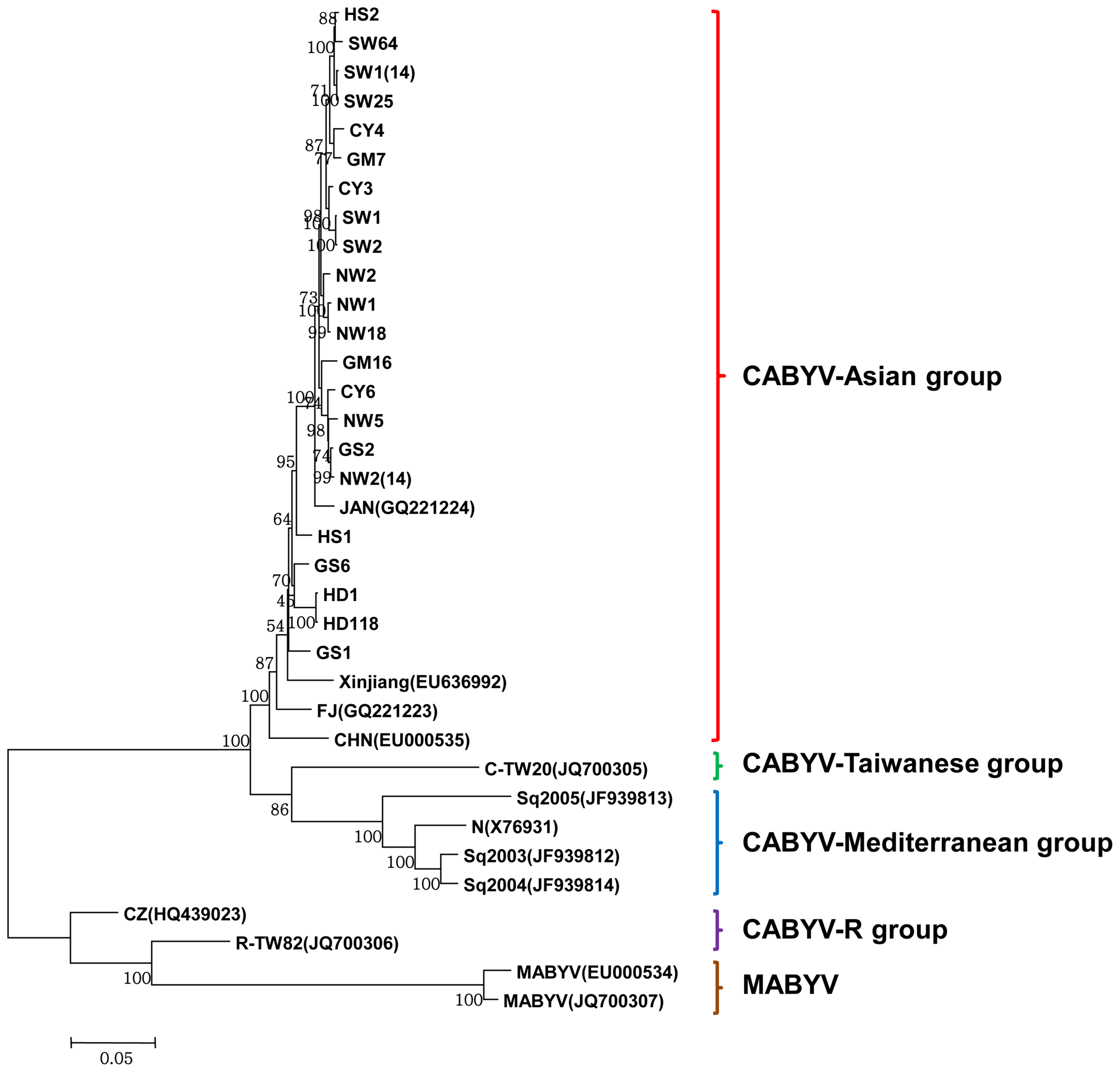
Phylogenetic trees reconstructed using the complete nucleotide sequences of the CABYV isolates. Phylogenetic trees were reconstructed using maximum likelihood in MEGA 6.
The reconstructed phylogenetic trees based on amino acid sequences of the six proteins (P0, P1, P1–P2, P3, P4, and P3–P5) and the nucleotide sequences of two non-coding regions (IR and 3′ UTR) showed that the Korean CABYV isolates clustered with the Asian group, similar to the tree based on nucleotide sequences (Supplementary Fig. 1 and 2). However, in the case of the 3′ proximal proteins and 3′ UTR, the Korean CABYV isolates were differentiated into two subgroups within the Asian group. In addition, the Chinese isolate CZ and Taiwanese isolate R-TW82, which belong to the CABYV-R group, grouped with MABYV based on full-length genome sequences and amino acids of 5′ proximal ORFs, but grouped with CABYV based on 3′ proximal ORFs. These results suggest that recombination occurred within CABYV isolates and between CABYV and MABYV isolates.
Sequence comparison
The nucleotide and amino acid sequence identities between CABYV isolates are summarized in Table 5. For the full-length genome nucleotide sequences, Korean CABYV isolates had 96–99% nt sequence similarity. CABYV-CY3, a Korean CABYV isolate, showed 94–98% nt sequence similarity with the Asian group including Japanese and Chinese isolates, 87–89% with the Mediterranean group, 88% with the Taiwanese group, 81–84% with the CABYV-R group, and 72% with the other polerovirus, MABYV.
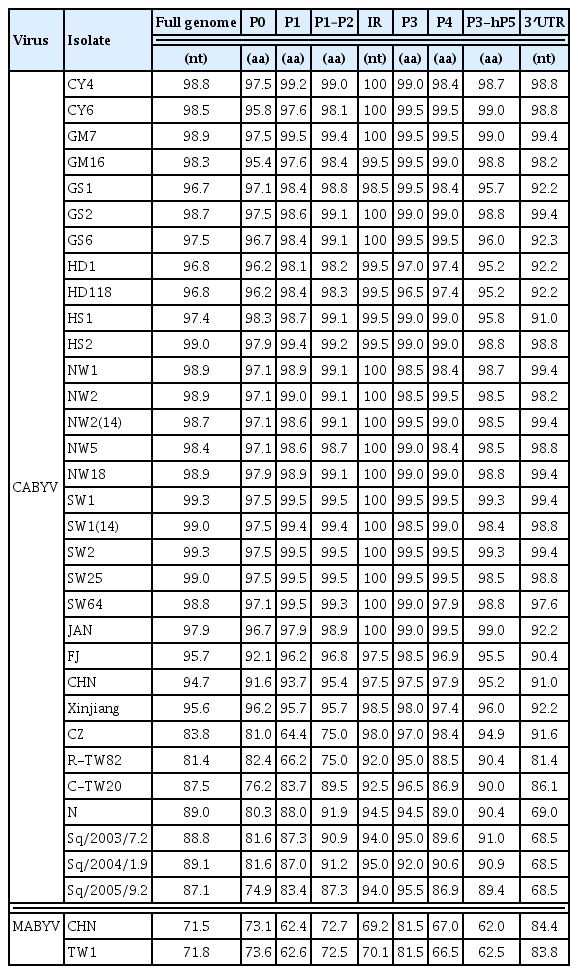
Nucleotide and amino acid sequence identities (%) between the Korean CABYV isolate CY3 and other CABYV isolates
Regarding the deduced amino acid sequences of six individual proteins, CABYV-CY3 (as a representative Korean CABYV isolate) showed relatively high sequence identity of 92–100% with the Asian group. In contrast, aa sequence identities between CABYV-CY3 and the Mediterranean group were 75–82% for P0, 83–88% for P1, 87–92% for P1–P2, 92–97% for P3(CP), 87–91% for P4(MP), and 89–91% for P3–P5. In comparison with CABYV-CZ and R-TW82, CABYV-CY3 had lower aa sequence identity of 65–75% for the 5′ proximal proteins (P1 and P1–P2), but 89–98% for the 3′ proximal proteins (P3, P4 and P3–P5). In addition, CABYV-CY3 shared only 62–82% aa sequence identity with MABYV isolates for each individual protein.
The nt sequence identities of the IR region and 3′ UTR were 92–100% and 69–92%, respectively, among the four CABYV groups, and were 70% and 84% with MABYV.
Recombination analysis
Recombination has been shown to significantly contribute to luteovirus diversity. To examine whether recombination events have occurred in the CABYV population, we aligned full-length nt sequences of 33 CABYV and 2 MABYV isolates using the Geneious method in Geneious Pro 8 and analyzed them using the RDP, GENECONV, BootScan, MaxChi, Chimaera, SiScan and 3Seq methods implemented in the RDP4 software with a highest acceptable P-value of 0.01. In total, 56 potential recombinant events were detected by at least one method; however, to reduce error, we included only recombination events supported by at least three methods. Using this criterion, 24 recombination events, including 12 recombination types, were detected in 17 CABYV isolates (Table 6). Among the Korean CABYV isolates, nine recombination events were detected in eight isolates of types 5, 7, 8, and 9. In particular, isolates HD1 and HD118 of recombination type 8 were detected as recombinants between the major parent HS2 and minor parent CZ. In this recombination event, the genomic region (nt 3,388-11) was replaced with the homologous region of CZ. This result could explain why these isolates belonged to the same group, which was distantly related to the other Korean CABYV isolates in the phylogenetic trees based on amino acid sequences of the 3′ proximal proteins (P3, P4, and P3–P5). The other recombinant isolates, GS1 and HS1, had the same parental isolates as the Chinese isolates, FJ and Xinjiang, with a type 8 recombination event, but their recombination was detected in the P3–P5 region (nt 4,601-4,893). These two regions, IR and P3–P5, were identified as hotspots for recombination of CABYV. On the other hand, as expected from phylogenetic and sequence analyses, the Chinese isolate CZ and Taiwanese isolate R-TW82 were reconfirmed as recombinants of CABYV and MABYV with P-values of 5.740 × 10−20 and 2.048 × 10−138, respectively.

Recombination in Korean CABYV populations
To further confirm the recombination, phylogenetic network analysis was performed using SplitsTree v. 4.1 program. The split decomposition analysis revealed that nine tentative recombinants formed a reticulate network structure (Fig. 5). Seven isolates except GM16 and C-TW20 were detected as recombinants by both RDP4 and SplitsTree v. 4.1 programs.
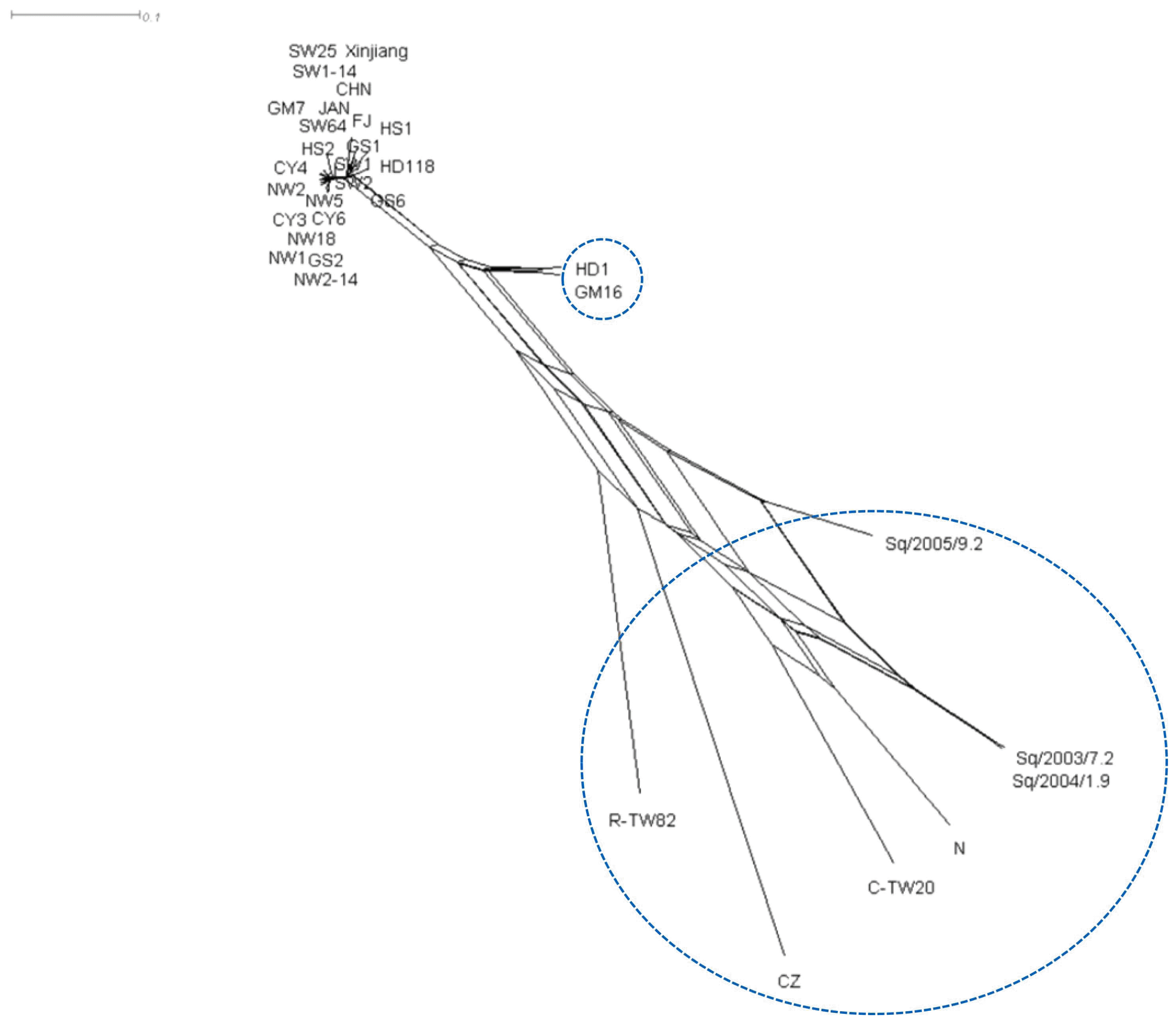
Split decomposition network of the CABYV population. Phylogenetic network analysis was performed using SplitsTree v. 4.1 program.
Discussion
Recently, CABYV was detected in Korea in melons showing yellowing symptoms using NGS and RT-PCR. Of 308 melon samples surveyed in seven areas during 2013–2014, 245 (80%) were positive for CABYV. To investigate the genomic structure, genetic diversity, and the possible origin of Korean CABYV population, we determined the full genome sequences of 22 CABYV isolates from CABYV-positive melon samples and analyzed their genetic diversity by comparison with the sequences of 11 CABYV isolates and 2 MABYV isolates as outgroups. The complete genomes of the Korean CABYV isolates range between 5,680 to 5,684 nt and encode six open reading frames (ORFs) that are separated into two regions by a non-coding internal region (IR) of 199 nt. The genomic organization of these isolates is typical of the genus Polerovirus. The deduced amino acid sizes of five proteins, the exception being the P3–P5 protein, were identical to those of the Asian CABYV group, which includes Japanese and Chinese isolates. Most Korean CABYV isolates had a P3–P5 protein of 668 aa, while some isolates comprised 667 aa due to the lack of one proline in the 5′ terminal region of P5.
Sequence comparison revealed that the Korean CABYV isolates shared 95–98% nt sequence identity and 92–100% aa sequence identities for six individual proteins with the Asian group (Table 5). In addition, the Korean CABYV isolates showed 82–89% nt sequence identity and 75–98% aa sequence identity with three other CABYV groups. Of the individual proteins, the 5′ proximal proteins were more variable than the 3′ proximal proteins. In particular, P0 was the most variable, while P3 (CP) was the most conserved. These characteristics; i.e., highly variable P0 and conserved P3 (CP), have been reported for other polerovirus species (Hauser et al., 2000; Huang et al., 2005; Xiang et al., 2010).
Using phylogenetic analyses based on full-length genome sequences, the Korean CABYV isolates clustered in the Asian group (Fig. 4). According to previous reports, CABYV isolates are divided into two groups that cluster geographically: the Asian and Mediterranean groups (Shang et al., 2009). However, our phylogenetic results suggest that CABYV isolates are divided into four groups: Asian, Mediterranean, Taiwanese, and R groups. Phylogenetic trees reconstructed using the amino acid sequences of six individual proteins and nt sequences of non-coding regions showed that Korean CABYV isolates consistently grouped into the Asian group. However, using 3′ proximal proteins and the 3′ UTR, the Korean CABYV isolates were classified into two subgroups within the Asian group. This shows the possibility of recombination in the IR region between the 5′ proximal and 3′ proximal proteins. Although the CABYV-R group belonged to CABYV, it grouped into MABYV in the 5′ proximal protein and 3′ UTR-based phylogenetic analyses, due to recombination between CABYV and MABYV isolates (Knierim et al., 2013).
To confirm the results of sequence and phylogenetic analyses, we investigated whether recombination occurred in the CABYV population using the RDP4 software. Twenty-four recombination events of 12 recombination types were detected in the analyzed CABYV population. Among them, nine recombination events occurred in the Korean CABYV isolates. Two recombination types in particular, 8 and 9, were detected in regions IR and the P3–P5 readthrough protein, respectively. These two regions have been reported as hotspots of RNA recombination in the family Luteoviridae, including CABYV (Gibbs and Cooper, 1995; Huang et al., 2005; Shang et al., 2009). In addition, recombination types 7 and 8 were detected as recombinants between HS2 as a major parent and CZ as a minor parent. This result could explain why the Korean CABYV isolates differentiated into two groups in phylogenetic trees based on the aa sequences of the 3′ proximal proteins (P3, P4, and P3–P5). Especially, some of CABYV isolates detected as tentative recombinants by RDP4 were consistently confirmed as recombinants by split decomposition analysis. Collectively, our results suggest that recombination is a major evolutionary force in the genetic diversification of the CABYV population in Korea.
In the present study, we analyzed the genetic diversity and structure of the CABYV population collected from melon plants. Our findings revealed that the Korean CABYV isolates belong to the CABYV-Asian group and that their genetic diversity is generated by recombination, as well as accumulation of mutations. Understanding the molecular characterization of viruses is essential for the development of strategies for the virus control.
CABYV, an important pathogen that causes yellowing symptoms in cucurbit crops, has been reported to infect nine cucurbit crops in China (Xiang et al., 2008a). In Korea, many cucurbit species are widely cultivated, and it has been confirmed that CABYV infected cucumber and oriental melon (Choi et al., 2015). Recently, we also could confirm that some watermelons and pumpkins showing mosaic and yellowing symptoms were co-infected with CABYV and other viruses including Watermelon mosaic virus or Zucchini yellow mosaic virus. CABYV became as one of the major viruses damaging cucurbits in Korea. However, knowledge of host range, pathogenicity, and vector transmission of Korean CABYV isolates is still limited. Further studies are needed for the aim of preventing the spread of CABYV.
Supplementary Information
Acknowledgements
This research was supported by a grant from the Agenda Program (PJ01130602), funded by the Rural Development Administration of Korea.