Introduction
Sugarcane (Saccharum spp.) is not only a key sugar-producing crop but is also a potentially renewable bioenergy crop that is grown in tropical and subtropical regions worldwide. The modern sugarcane cultivars are a complex of aneupolyploids that result from commercial hybrid breeding programs involving several species and consist of approximately 80% S. officinarum, 10-15% S. spontaneum and 5-10% recombinant chromosomes (DŌĆÖHont et al. 1996). The crop is subjected to various stresses caused by biotic pathogens and abiotic conditions during the growing period. Sugarcane bacilliform viruses (SCBVs) that belong to the genus Badnavirus (family Caulimoviridae) are one of the main species that infect sugarcane. In 1985, SCBV was first identified in the infected sugarcane cultivar B34104 in Cuba, and then spread into sugarcane-growing regions worldwide (Autrey et al., 1995; Lockhart et al., 1996). SCBV is naturally transmitted by insect vectors, the mealybug species Saccharicoccus sacchari and Dysmicoccus boninsis, and can also be experimentally transmitted by crude virus-containing sap or via Agrobacterium-mediated inoculation (Lockhart et al., 1996). In addition, long-distance transport of SCBV-infected cane materials is an important mean of virus dissemination. The natural host range of SCBV is not limited to sugarcane, but also includes other grasses such as Sorghum halepense, Brachiaria sp., Panicum maximum, and Rottboellia exaltata; artificial SCBV infection succeeded in Oryza sativa, Musa sp., and Sorghum vulgare (Bouhida et al., 1993; Lockhart et al., 1996; Viswanathan et al., 1996). The visual symptoms of SCBV infection in cultivated sugarcane are stunted growth, flecking, and chlorotic stripes, but depend on the combination of host clones and virus strains (Comstock and Lockhart, 1990; Lockhart et al., 1996; Viswanathan et al., 1996). SCBV infection may lead to significant losses in biomass production in susceptible sugarcane cultivars (Lockhart and Autrey, 2000).
SCBVs that infect sugarcane are a genetically heterogeneous Badnavirus species complex. At present, four badnaviruses, Sugarcane bacilliform Guadeloupe A virus (SCBGAV), Sugarcane bacilliform Guadeloupe D virus (SCBGDV), Sugarcane bacilliform MO virus (SCBMOV), and Sugarcane bacilliform IM virus (SCBIMV), which originally clustered in the SCBV-A, SCBV-D, SCBV-E, and SCBV-F phylogenetic groups/genotypes, respectively, have been assigned by the International Committee on Taxonomy of Viruses (ICTV) as different species in the Badnavirus genus (Adams and Carstens, 2012; Adams et al., 2016; Geering and Hull, 2012). A total of 12 full length genomes were obtained for global SCBV isolates from Morocco (SCBMOV-MOR) (Bouhida et al., 1993), Australia (SCBIMV-QLD) (Geijskes et al., 2002), Guadeloupe (SCBGAV-R570, SCBGAV-R51129, and SCBGDV-Batavia) (Muller et al., 2011), India (SCBV-BT, SCBV-BRU, SCBV-BO91, SCBV-Iscam, and SCBV-BB) (Karuppaiah et al., 2013), and China (SCBV-CHN1 and SCBV-CHN2) (Sun et al., 2016). The full SCBV genome is approximately 7.3-8.0 kilobase pair (kb) in size and consists of circular double-stranded DNA that replicate through a virus-encoded reverse transcriptase (RT). SCBV retains the Badnavirus genome organization which consists of three open reading frames (ORFs) on the positive strand; ORFs 1 and 2 encode two small proteins of 176-185 and 122-135 amino acids (aa), respectively, but the function of these proteins are unknown (Geering and Hull, 2012; Sun et al., 2016). ORF 3 encodes a polyprotein of 1786-1933 amino acids which includes several functional units, such as a movement protein, aspartic protease, coat protein, RT, and ribonuclease H (RNase H) proteins (Bouhida et al., 1993; Geering and Hull, 2012; Geijskes et al., 2002).
Various PCR assays have been used to screen for badnaviruses with three previously-developed primer pairs, SCBV-F5/SCBV-R5 (Braithwaite et al., 1995), Badna-FP/Badna-RP (Yang et al., 2003), and SCBV-F/SCBV-R (Wu et al., 2016), targeted at the RT/RNase H coding region. The three primer sets have been used in SCBV detection assays and the resulting PCR products were cloned and sequenced to explore SCBV genetic diversity (Braithwaite et al., 1995; da Silva et al., 2015; Karuppaiah et al., 2013; Muller et al., 2011; Wu et al., 2016). Currently, 18 phylogenetic groups (genotypes) of SCBV have been reported worldwide (SCBV-A to SCBV-R), and they are to some extent associated with geographical regions. Five SCBV genotypes, SCBV-A, SCBV-B, SCBV-C, SCBV-D, and SCBV-G, are present in France (Muller et al., 2011), seven SCBV genotypes, SCBV-E, SCBV-H, SCBV-I, SCBV-J, SCBV-K, SCBV-L, and SCBV-M, have been detected in India (Karuppaiah et al., 2013; Rao et al., 2014), and five SCBV genotypes, SCBV-A, SCBV-C, SCBV-F, SCBV-M, and SCBV-H, have been found in the Brazilian sugarcane germplasm collection (da Silva et al., 2015). In China, eight SCBV genotypes (SCBV-G, SCBV-H, SCBV-L, and from SCBV-N to SCBV-R) have been reported, and the two prevalent genotypes (SCBV-Q and SCBV-R) were found in sugarcane planting regions (Wu et al., 2016).
Some previous reports have described the occurrence and prevalence of SCBVs in China (Cai et al., 2009; Li et al., 2010; Wu et al., 2016; Xu et al., 2015); in the present study, two novel SCBV genotypes are proposed and two SCBV species are reported for the first time in China. Our findings will contribute to a lager understanding of the genetic diversity of SCBVs, and will also aid in the screening of sugarcane germplasm to minimize the spread of viral variants across geographical boundaries via contaminated plant material.
Materials and Methods
Leaf tissue collection
A total of 392 sugarcane leaf samples were collected from three sources; vegetative cuttings exchanged for national sugarcane regional tests in Fuzhou, Fujian province, varieties in commercial fields in Yunnan province, and a wild germplasm collection in Hainan province, China from 2015 to 2017. Initially, 114 leaf samples (three randomly-chosen leaves from each clone) were collected from 38 sugarcane clones that were originally introduced from different Chinese sugarcane research institutes into Fuzhou, Fujian province in 2015. The second set of 216 leaf samples (six leaves per clone) were collected from the wild germplasm collection in Sanya, Hainan province in 2016, and the third set of 62 leaf samples (one leaf per cultivar in a field) was collected from three sugarcane planting regions (Baoshang, Dehong, and Lincang) in Yunnan province in 2017. The leaf samples are described in Supplementary Table 1. The leaf samples were rinsed with 75% ethanol and stored at ŌłÆ80┬░C until they were used for DNA extraction.
DNA isolation and PCR amplification
Total DNA was extracted from sugarcane leaf samples using the cetyltrimethylammonium bromide (CTAB) method (Haible et al., 2006). A set of degenerate primers SCBV-F and SCBV-R were used to amplify the RT/RNase H genomic region of SCBV (Wu et al., 2016). Total leaf DNA (200 ng/╬╝l) was used as a template for PCR amplification with EX Taq polymerase (TaKaRa, Dalian, China) under the following conditions: an initial denaturation step at 94┬░C for 5 min, followed by 35 cycles of 94┬░C for 30 s, 58┬░C for 30 s, 72┬░C for 45 s, and a final extension step at 72┬░C for 10 min. Amplified DNA was electrophoresed on 1.5% agarose gels, stained with ethidium bromide, and then visualized with a ChemiDoc XRS instrument (Bio-Rad Laboratories, Hercules, CA, USA) to confirm that the amplified DNA fragments were of the expected size of 726 base pair (bp).
Cloning and sequencing of PCR products
All amplified DNA fragments obtained from each leaf sample were purified using the E.Z.N.A. Gel Extraction Kit (Omega Bio-Tek Inc., Georgia, USA) and then cloned into the pMD19-T vector (TaKaRa, Dalian, China) following the manufacturerŌĆÖs instructions. The ligation reactions were used to transform competent E. coli DH5╬▒ cells (Tiangen Biotech Co. Ltd., Beijing, China). Three positive colonies per PCR product were randomly selected and the nucleotide (nt) sequence was determined in both directions by Shanghai Shenggong Biology Company (China). The sequence contigs of the three colonies from each leaf DNA sample were compared among themselves using DNA-MAN (version 8). An accurate ŌĆśuniqueŌĆÖ sequence was obtained when the sequences of the three colonies from each leaf sample showed Ōēź 99% similarity.
DNA sequence alignment and analysis
One-hundred eleven DNA sequences of 726 bp in length were obtained in this study. Thirty-four reference sequences that are representative of 18 different worldwide SCBV genotypes (Wu et al., 2016) were downloaded from the NCBI GenBank database. All the DNA sequences were trimmed at both ends to a length of 480 nucleotides to accommodate a considerable number of GenBank sequences in the analyses, and then were subjected to multiple alignments using CLUSTALW as implemented in MEGA6 (Tamura et al., 2013).
Recombination events were detected in the dataset of aligned RT/RH sequences with seven algorithms implemented in RDP v.3.44 software (Martin et al., 2010) using the default parameters. The neighbor-joining (NJ) phylogenetic tree, which included a corresponding sequence from an isolate of Commelina yellow mottle virus (ComYMV, GenBank no. NC001343) as an outgroup in the alignment, was constructed with the MEGA6 program. The robustness of the trees was determined via bootstrap analysis (1,000 replicates), and the bootstrap values were included at the internodes on the tree (Felsenstein, 1985). Pairwise sequence comparison (PASC) was carried out with the MEGA6 (Tamura et al., 2013) and BioEdit programs to estimate the genetic distances and pairwise identities of the nucleotide and amino acid sequences, respectively.
Results
PCR detection of SCBV in leaf samples
Of all the 392 sugarcane leaf samples, 16.1% (63/392) were scored as positive for SCBV with the SCBV-F/SCBV-R primer pair; 38.6% (44/114) of the Saccharum spp. hybrid varieties from Fujian, 8.1% (5/62) of the Saccharum spp. hybrid varieties from Yunnan, and 6.5% (14/216) of the wild germplasm accessions from Hainan (Table 1). Only five of 36 wild germplasm clones were detected as positive in Striped Cheribon (S. officinarum), Sewari (S. robustum), Yacheng11 (S. spontaneum), Guangdong29 (S. spontaneum), and Hainan92-79 (Erianthus arundinaceus). All PCR products amplified from SCBV-positive leaf DNA samples were cloned and sequenced. The SCBV sequences obtained in this study were submitted to the NCBI GenBank database (Supplementary Table 1).
Phylogenetic analysis
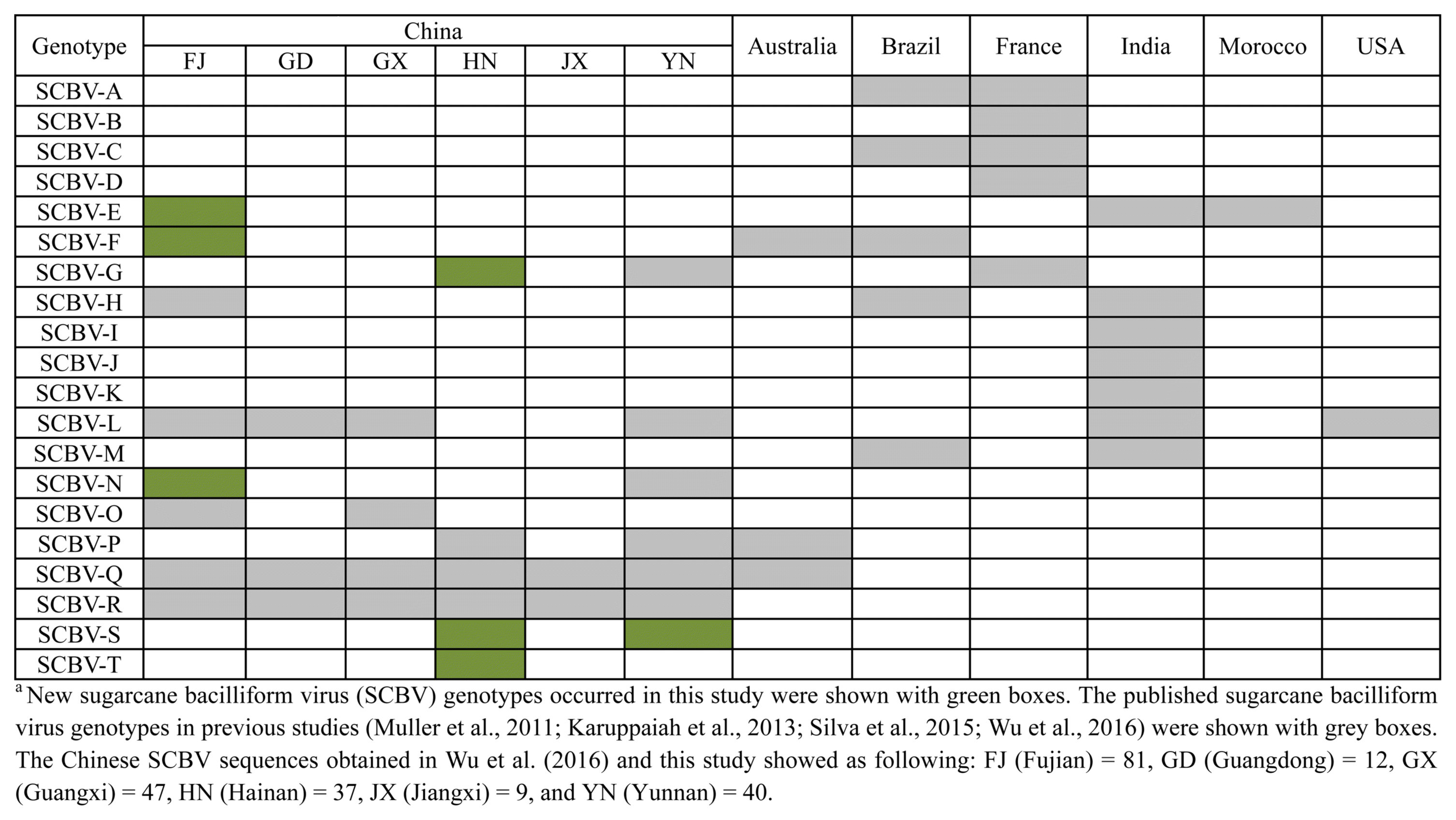
Because recombination may confound virus phylogenetic analyses, the aligned nucleotide sequences were subjected to recombination analysis to identify conflicting phylogenetic signals. No significant recombination events were detected using seven algorithms. Thus, the dataset of SCBV sequences and one sequence of ComYMV was used for phylogenetic analysis. The neighbor-joining phylogenetic tree revealed that all of the sequences in this study could be classified into 20 SCBV genotypes (from SCBV-A to T) including the 18 previously-published SCBV genotypes (Fig. 1). We identified two novel SCBV genotypes, SCBV-S and SCBV-T, in this study. Twenty-four and 32 sequences from the Fuzhou isolates clustered into the SCBV-E and SCBV-F groups, respectively, and the reminding 11 sequences clustered into the SCBV-L, -N, and -Q groups. Thirteen sequences from the Yunnan isolates clustered into four groups (SCBV-L, SCBV-Q, SCBV-R, and SCBV-S). Notably, 27 sequences from the Hainan isolates infecting S. robustum, S. spontaneum, and E. arundinaceus were clustered into the two newly identified groups called SCBV-S and SCBV-T in our study, whereas the four isolates (HNSo1, HNSo2, HNSo3, and HNSo4) from S. officinarum clustered with the SCBV-G group isolates (Fig. 1). The geographical distribution of SCBV genotypes from this study and previous reports in China and other countries is shown in Fig. 2. Apart from SCBV-S and SCBV-T, another two SCBV groups (SCBV-E and SCBV-F) that were assigned to the Badnavirus species SCBMOV and SCBIMV by ICTV, respectively, are reported for the first time in China. Besides, SCBV-G and SCBV-N groups were firstly reported in Hainan and Fujian provinces, China, respectively.
Pairwise sequence identity between the SCBV groups
Pairwise sequence identity between the 20 phylogenetic groups (from SCBV-A to SCBV-T) ranged from 50.6-93.2% and 53.0-99.3% at the nucleotide and amino acid levels, respectively (Table S2). The SCBV-S and SCBV-T groups shared sequence identities of 90.0-93.2% (nt) and 92.7-96.9% (aa). In addition, SCBV-S shared 57.6-92.2% (nt) and 53.0-99.3% (aa) identity, whereas SCBV-T shared 58.4-90.0% (nt) and 53.0-96.9% (aa) identity with the other 18 SCBV genotypes (Table S2). The nt sequences of the Fuzhou (Fujian province) isolates shared 75.0-100% identity with each other and shared 59.8-62.4%, 59.4-62.4%, 77.0-99.8%, and 76.2-99.6% identity with four ICTV-recognized Badnavirus species isolates, SCBGAV, SCBGDV, SCBMOV, and SCBIMV, respectively (Table 2). The Yunnan province isolates shared 86.4-100% nt identity with one another and 59.2-61.2%, 59.2-61.4%, 76.8-79.2%, and 87.4-90.6% identity with the SCBGAV, SCBGDV, SCBMOV, and SCBIMV isolates, respectively. The Hainan province isolates shared 86.8-100% nt identity with each other and had nt identities of 58.4-61.4%, 59.4-61.6%, 76.6-79.8%, and 88.2-90.6% with the SCBGAV, SCBGDV, SCBMOV, and SCBIMV isolates, respectively.
Pairwise nucleotide sequence genetic distances between the SCBV groups
Pairwise genetic distances between the 20 phylogenetic groups ranged from 0.07-0.67 (nt) and 0-0.64 (aa) (Table S3). The genetic distance between SCBV-S and SCBV-T ranged from 0.07-0.11 and 0.03-0.08 at the nt and aa levels, respectively. SCBV-S showed genetic distances of 0.08-0.66 (nt) and 0.01-0.64 (aa), whereas SCBV-T showed distances of 0.11-0.63 (nt) and 0.03-0.64 (aa) with the other 18 SCBV genotypes (Table S3). Genetic distances between the individual Fuzhou isolates ranged from 0.00 to 0.31 (nt), while the distances between the Fuzhou isolates and the SCBGAV, SCBGDV, SCBMOV and SCBIMV isolates were 0.52-0.60, 0.52-0.59, 0.00-0.28, and 0.00-0.29, respectively (Table 2). The Yunnan province isolates showed 0.00-0.15 nt genetic distance with each other, and showed genetic distances of 0.55-0.60, 0.54-0.59, 0.25-0.29, and 0.10-0.14, respectively, with the SCBGAV, SCBGDV, SCBMOV, and SCBIMV isolates. Genetic distances within the Hainan province isolates were 0.00-0.15 (nt), and were 0.55-0.62, 0.54-0.58, 0.24-0.29, and 0.10-0.13 between the Hainan isolates and SCBGAV, SCBGDV, SCBMOV, and SCBIMV, respectively (Table 2).
Discussion
Sugarcane is extensively cultivated worldwide, and is both an important sugar-producing crop and biofuel feedstock. The global exchange of germplasm has occurred frequently in an effort to broaden the genetic base of the pool of sugarcane hybrid parents, but this has also increased the risk of long-distance virus transmission across geographical barriers resulting from the transport of SCBV-infected vegetative cuttings. Sugarcane-infecting badnaviruses present significant genomic nucleotide sequence variability and serological heterogeneity (Lockhart et al., 1996; Sun et al., 2016). Hence, in this study, we further investigated the genetic diversity of the genetically heterogeneous SCBV complex based on three sets of sugarcane leaf samples from Fujian, Yunnan, and Hainan provinces in China, even though several studies have already reported the existence of diverse SCBVs infecting sugarcane in China (Li et al., 2010; Wu et al., 2016; Xu et al., 2015).
The RT/RNase H coding region is a common taxonomic marker for species demarcation within the genus Badnavirus (Geering and Hull, 2012). The demarcation criterion is > 20% nt variation in the RT/RNase H region, which was proposed by the ICTV (Geering and Hull, 2012). Also, the threshold for nt genetic distance (> 0.2) in the RT/RNase H region between species in the Badnavirus genus is another demarcation criteria (Bousalem et al., 2008). Genetic variation and genetic distance within the RT/RNase H regions of the two novel SCBV groups, SCBV-S and SCBV-T, were compared with the representative isolates of four Badnavirus species, SCBGAV, SCBGDV, SCBIMV-QLD, and SCBMOV, and showed that the SCBV-S and SCBV-T groups had > 20% nt variation when compared with SCBMOV, SCBGAV, and SCBGDV, but had only 9.4-11.8% nt variation compared to SCBIMV. Similarly, the nt genetic distances between the SCBV-S and SCBV-T group isolates were > 0.2 when compared with the four SCBV species except for SCBIMV. It is noteworthy that the SCBV-N isolate group met both of the criteria for species in the Badnavirus genus, indicating that SCBV-N may well be a novel SCBV species (Wu et al., 2016). The two SCBV species SCBMOV (SCBV-E group) and SCBIMV (SCBV-F group) were assigned in 2012 (Adams and Carstens, 2012), and the other SCBV species SCBGAV (SCBV-A group) and SCBGDV (SCBV-D group) were assigned more recently in 2016 by ICTV (Adams et al., 2016). In addition, more than four SCBV species, including SCBV-C in Guadeloupe (Muller et al., 2011), SCBV-H, SCBV-I, and SCBV-J in India (Karuppaiah et al., 2013) have been proposed. However, formal recognition of these Badnavirus SCBV species is still pending.
In our previous studies, five genotypes of SCBV (SCBV-H, SCBV-L, SCBV-O, SCBV-Q, and SCBV-R) were reported in Fujian province, six genotypes (SCBV-G, SCBV-L, SCBV-N, SCBV-P, SCBV-Q, and SCBV-R) in Yunnan province, and three genotypes (SCBV-P, SCBV-Q, and SCBV-R) in Hainan province (Sun et al., 2016; Wu et al., 2016). However, in this study we report nine SCBV genotypes in Fujian, Yunnan, and Hainan provinces. Of the nine SCBV genotypes, we reported for the first time that SCBMOV (SCBV-E) and SCBIMV (SCBV-F) occur in China, and that SCBV-N is present in Fujian province. The two SCBV species (SCBMOV and SCBIMV) and the one genotype (SCBV-N) may have been imported from other Chinese provinces because these leaf samples were collected from sugarcane clones that were introduced into Fuzhou from other provinces as SCBV-infected vegetative cuttings imported for national sugarcane regional tests during 2013-2015. Another possibility is that they could have been infected by existing SCBVs in Fuzhou during the cultivation period. Also, the two novel genotypes (SCBV-S and SCBV-T) and group SCBV-G that once occurred in Yunnan province were detected in the wild germplasm collection in Hainan province for the first time. Most of the sequences from the wild germplasm clones clustered with the two newly-identified groups SCBV-S and SCBV-T in our study. Exceptions were HNSo1, HNSo2, HNSo3, and HNSo4, which infect S. officinarum and were included in the SCBV-G group, suggesting that host plants could be an important factor for SCBV genetic differentiation.
Highly divergent SCBV variants have been reported worldwide. Genetic recombination, as an evolutionary force, may have contributed to the emergence of genetic diversity in SCBV. Stronger evidence of recombination was detected within and between SCBV groups in the RT/ RNase H gene coding region (Wu et al., 2016), and also at the genome level (Sun et al., 2016), and was even detected between SCBV and banana streak virus (BSV) (Sharma et al., 2015). However, only very weak recombination signals were detected in some SCBV sequences analyzed in this study. It has been reported that SCBV can naturally or experimentally infect banana, which is a natural host plant for BSV (Bouhida et al., 1993). Thus, it is possible that recombination events between SCBV and BSV could have occurred prior to the time of divergence from the last common ancestor of the currently-known banana and sugarcane badnaviruses (Sharma et al., 2015). It is notable that some SCBV genotypes share a high degree of similarity with some BSV species (Iskra-Caruana et al., 2014; Muller et al., 2011; Wu et al., 2016). In addition to recombination, repeated bottleneck events such as horizontal vector-mediated transmission, cell-to-cell movement, and systemic spread within the host plant could also contribute to dynamic SCBV populations (Wu et al., 2016). All of the published studies suggest a complicated phylogenetic makeup of SCBV isolates worldwide, and also show that SCBV isolates are extremely diverse.
The results presented here provide a more complete picture of the prevalence, distribution, and genetic diversity of the SCBV species complex in China. In addition, our findings will play a key role in understanding the population structure of SCBV in semi-perennial and perennial host plants, and will also aid in the screening of sugarcane germplasm worldwide to reduce the spread of genetic variants of SCBV via contaminated plant material. With the continued discovery of new SCBV genotypes, it is important that the phylogenetic groups (genotypes) be identified to the level of species or strains in the genus Badnavirus.
 PDF Links
PDF Links PubReader
PubReader Full text via DOI
Full text via DOI Full text via PMC
Full text via PMC Download Citation
Download Citation Supplement
Supplement Print
Print