Wisteria Vein Mosaic Virus Detected for the First Time in Iran from an Unknown Host by Analysis of Aphid Vectors
Article information

Abstract
The development of reverse transcription–polymerase chain reaction using degenerate primers against conserved regions of most potyviral genomes enabled sampling of the potyvirome. However, these assays usually involve sampling potential host plants, but identifying infected plants when they are asymptomatic is challenging, and many plants, especially wild ones, contain inhibitors to DNA amplification. We used an alternative approach which utilized aphid vectors and indicator plants to identify potyviruses capable of infecting common bean (Phaseolus vulgaris). Aphids were collected from a range of asymptomatic leguminous weeds and trees in Iran, and transferred to bean seedlings under controlled conditions. Bean plants were tested serologically for potyvirus infections four-weeks post-inoculation. The serological assay and symptomatology together indicated the presence of one potyvirus, and symptomology alone implied the presence of an unidentified virus. The partial genome of the potyvirus, encompassing the complete coat protein gene, was amplified using generic potyvirus primers. Sequence analysis of the amplicon confirmed the presence of an isolate of Wisteria vein mosaic virus (WVMV), a virus species not previously identified from Western Asia. Phylogenetic analyses of available WVMV sequences categorized them into five groups: East Asian-1 to 3, North American and World. The Iranian isolate clustered with those in the World group. Multiple sequence alignment indicated the presence of some genogroup-specific amino acid substitutions among the isolates studied. Chinese isolates were sister groups of other isolates and showed higher nucleotide distances as compared with the others, suggesting a possible Eastern-Asian origin of WVMV, the main region where Wisteria might have originated.
About 80% of known plant viruses are insect-transmissible, and more than 70% of all insect-borne viruses are transmitted by hemipterans (Fereres and Raccah, 2015), including aphids. Aphids belong to the superfamily Aphidoidea, and over 5,000 species have been recorded (Foottit et al., 2008). While some aphids have very narrow host ranges, being specialized to feed from plants of one species or genus, others feed across multiple families. For example, Myzus persicae (green peach aphid) colonizes plants from over 50 families. The combination of feeding behavior, sexual and asexual life cycles, and ability to transmit many viruses make aphids very serious pests of agriculture in temperate regions. Aphids transmit plant viruses, especially single-stranded RNA viruses, through non-circulative (non-persistent or semi-persistent) and circulative (propagative or non-propagative) strategies; most aphid-borne viruses are transmitted non-persistently (Guerrieri and Digilio, 2008; Hull, 2014).
Human activities, such as the international trade in fresh plant produce and propagules, and farming activities at the interfaces of managed and wild systems, enhance the possibility of viruses expanding their geographical and host ranges, leading to emergence of new viruses and epidemics (MacDiarmid et al., 2013; Wylie et al., 2014). Hence, constant monitoring and diagnosis of plant viruses is important. Aphid-transmitted plant viruses have mainly been detected directly from host plants rather than from aphid vectors (e.g., Farzadfar et al., 2002). Some plants, especially wild ones, contain inhibitors (such as polysaccharides, starches, polyphenols, etc.) that may restrict effectiveness of RNA extraction and subsequent enzymatic amplification steps (Schrader et al., 2012), and affect the efficiency of mechanical transmission trials as well (Hull, 2014). On the other hand, aphids and widely-used virus indicator plants generally lack these inhibitors.
Studies of non-aphid vectors, including ticks, mosquitoes, and whiteflies, have revealed novel viruses and others not previously known to exist in the study area (Atoni et al., 2018; Donaldson et al., 2010; Islam et al., 2018; Pettersson et al., 2017). Other studies have focussed on animals that are not virus vectors themselves, but consume virus vectors. For example, the guano of bats that consume insect vectors of plant viruses contains plant viruses (Li et al., 2010). Few studies have been performed using aphids to screen for vectored viruses. These studies have resulted in finding viruses that infect aphids themselves rather than those that infect plants (Feng et al., 2017; Wamonje et al., 2017). Direct virus sampling of aphids from host plants or by trapping/collecting aphids followed by reverse transcription–polymerase chain reaction (RT-PCR) or high-throughput sequencing (HTS), are some potential methods for detecting and studying aphid-borne plant viruses. One review described direct detection of viruses from their vectors as a “major challenge to diagnosticians and plant pathologists” (Mumford et al., 2006). A potential disadvantage of such approaches is the difficulty of subsequent establishment of virus cultures for further studies from extracts of single aphids, when a virus is diagnosed. By enabling captured aphids to transmit any viruses they are carrying to indicator plants, a virus culture is thereby established. A similar method has been previously utilized to assess efficiency of transmission of specific viruses, e.g., potyviruses (Harrington et al., 1986).
The Potyviridae is the largest family of plant-infecting RNA viruses, and many species are aphid-borne (Wylie et al., 2017). Potyviruses are very important plant viruses economically and are widespread in cultivated plants throughout the world, and are found infecting wild plants as well (Roossinck, 2012). Several sets of degenerate primers exist that detect conserved regions of potyvirus genomes through RT-PCR tests (Ha et al., 2008; Pappu et al., 1993; Zheng et al., 2010), and thereby provide information about the identities of most potyviruses in the region. Seven potyviruses of legumes were previously reported in Iran. These include bean common mosaic virus (BCMV), bean common mosaic necrosis virus (BCMNV), bean yellow mosaic virus (BYMV), cowpea aphid-borne mosaic virus (CABMV), pea seed-borne mosaic virus (PSbMV), peanut mottle virus (PeMoV), and soybean mosaic virus (SMV) (Esfandiari et al., 2006; Golnaraghi et al., 2004; Kaiser et al., 1968; Shahraeen et al., 2005); all of these viruses were detected directly from their host plants.
In the current work, aphid vectors and indicator plants were utilized to sample the ‘potyvirome’, a collective term for all potyvirus genomes in the area, particularly those capable of infecting common beans in Iran. Potyvirus detection was performed by serological assays using broad-spectrum antibodies and degenerate RT-PCR primers, followed by sequencing and sequence analyses. The results clearly confirmed the natural occurrence of a new potyvirus, i.e., Wisteria vein mosaic virus (WVMV), in the country. We compared the sequences from Iran and other geographical regions for the evolutionary characterization of the virus. Our studies, to our knowledge, provide the first phylogenetic analysis of WVMV isolates.
Materials and Methods
Aphid collection, aphid-transmission, and serological assays
In June 2014, live colonizing (apterous) aphids were collected from wild asymptomatic leguminous herbs and trees in Tehran province, one of the main pulse-producing provinces of Iran. The location of each sample was recorded by Global Positioning System (GPS). Eleven aphid samples consisting of 10–20 insects per sample were immediately transferred to young healthy bean (Phaseolus vulgaris) seedlings, grown from seeds under greenhouse conditions, for an inoculation access period of at least 1 h. Inoculation of bean plants was performed at cotyledon to two-leaf growth stages under field conditions, and the plants were then covered. Inoculated plants were transported to an insect-proof greenhouse. Aphids were preserved in 70% ethanol for later identification by A. Rezvani (Iranian Plant Protection Research Institute, Tehran, Iran). Potyvirus infections of the inoculated plants were tested by plate-trapped antigen enzyme-linked immunosorbent assay (Mowat and Dawson, 1987) 28 days post-inoculation using Poty group test kit (Bioreba, Reinach, Switzerland) according to the manufacturer’s instructions. OD values were measured at 405 nm using a microtiter plate reader (EL800, Bio-Tek Instruments, Winooski, VT, USA). A sample was considered virus-positive if its OD was equal to or exceeded three times the mean value of the negative controls (healthy) one hour after adding the substrate (p-nitrophenyl phosphate).
Total RNA extraction and RT-PCR
Isolate Th-W2259 was studied further. To this end, partial genomic sequence of the potyvirus isolate was determined. First, total RNA was extracted from 0.02–0.03 g of dried infected bean leaves using a LiCl-based protocol (de Fátima Rosas-Cárdenas et al., 2011). We used 5 μl of DNase I (50 u/μl; Thermo Scientific, Waltham, MA, USA) to remove DNA from the total RNAs obtained; the enzyme was then deactivated by adding 10 μl EDTA (50 mM; Thermo Scientific) and incubation at 65°C for 10 min. Total RNA was finally precipitated, resuspended in RNase/DNase free water and stored at −20°C. First-strand cDNA synthesis was performed using M-MuLV reverse transcriptase (CinnaGen, Tehran, Iran), according to the manufacturers’ instructions. PCR amplifications were done using SmarTaq DNA polymerase (CinnaGen), and thermocycling was carried out in a MyCycler thermal cycler (Bio-Rad, Hercules, CA, USA). All primers used in this study were synthesized by Metabion (Martinsried, Munich, Germany) or CinnaGen.
RT-PCR tests were conducted using universal cylindrical inclusion (CI), nuclear inclusion body protein (NIb), and NWCIEN primers according to previously published procedures (Ha et al., 2008; Valouzi et al., 2017; Zheng et al., 2010). To obtain the complete coat protein (CP) gene sequence of the isolate, two new primers (WVNIb1P: 5′-TTGTTTGCCCTTGGACAGTG-3′ and WVPseq: 5′-ATGTATGCTTCAGCCGCATC-3′) were designed based on the partial NIb and CP gene sequences of the isolate, respectively. These primers were then used to amplify the gap between the NIb and CP regions; the PCR program was 94°C for 4 min, 38 cycles of 94°C for 1 min, 50°C for 45 s and 72°C for 2 min, and finally 72°C for 10 min. PCR products and DNA ladder (Thermo Scientific) were separated by agarose gel electrophoresis, visualized using GelRed staining (Biotium Inc., Fremont, CA, USA), and photographed with ultraviolet-illumination.
Sequencing
To determine the nucleotide sequence of PCR products, fragments of expected sizes were excised from the gel and cleaned by the sequencing service of Bioneer (Daejeon, Korea). Sanger sequencing was carried out in both directions using the amplification primers. Pseq primer (Valouzi et al., 2017) was used for sequencing of the PCR product obtained using NWCIEN primers. Sequences of each region were determined using two independent PCR products. The sequences of adjacent regions were overlapped by about 100 bp to ensure that they were from the same genome and were not from different components of a genome mixture (Farzadfar et al., 2009). Bio-Edit software version 7.2.5 (Hall, 1999) was utilized for trimming and assembling of nucleotide sequence data.
Sequence analysis
As a first step, Basic Local Alignment Search Tool (BLAST) (Altschul et al., 1997) was applied to search GenBank databases (available at the National Center for Biotechnology Information, NCBI; https://www.ncbi.nlm.nih.gov/) to identify the sequences that most closely matched the nucleotide and deduced amino acid sequences obtained in this study. In the second step, the partial sequences of the CI and NIb genes and the complete sequence of the CP gene of the isolate Th-W2259 were concatenated (2,277 nts length). This sequence and the corresponding regions of 78 reference sequences from a set of representative species from the genus Potyvirus (ICTV) (Wylie et al., 2017) were used in phylogenetic analysis to confirm the results of database searches. A sequence from an isolate of ryegrass mosaic virus (RGMV) (accession no. NC_001814), genus Rymovirus, was used as the outgroup (Zheng et al., 2010) (Supplementary Table 1). The sequences were first aligned and degapped (Farzadfar et al., 2009). Evolutionary relationships between the viral sequences were deduced using maximum likelihood (ML) and neighbor-joining (NJ) algorithms in Mega 6.0 software (Tamura et al., 2013). ML analysis was done using the ‘find best substitution model’, i.e., general time-reversible model with a gamma distribution of site-rate variants across four categories and a proportion of invariant sites. The ML trees were found by subtree pruning and regrafting trees. We also did NJ analysis using Kimura two-parameter model. Both methods were done using 1,000 bootstraps. Trees were graphically edited using Interactive Tree of Life (iTOL) v4 (Letunic and Bork, 2019): nodes with less than 50% bootstrap support in both methods were collapsed and the joining branches of a collapsed node were adjusted to include the length of the collapsed branch and maintain most patristic distances (Zheng et al., 2010).
Sequence analysis of WVMV isolates
The CP gene sequence of WVMV isolate from Iran and 13 other isolates from the GenBank sequence database were used (Table 1). The results of BLAST searches showed that the CP sequence of an isolate of SMV, GenBank accession no. FJ640971, closely matched that of the WVMV sequences, and this sequence was used for the outgroup. The sequences were aligned and degapped, with or without the SMV sequence (Farzadfar et al., 2009). The recombination detection programs in RDP 4.39 software (Martin et al., 2015) were used to predict presence of recombination events in the aligned sequences. A sequence was considered “recombinant” if the recombination event was detected by at least three methods (P < 1 × 10−6). These procedures were performed according to the earlier recommendations (Farzadfar et al., 2009; Posada, 2002). Phylogenetic analysis was performed by Mega 6.0 software using the SMV sequence as the outgroup, as described above, except that the best nucleotide substitution models used for ML analysis were Tamura three-parameter model with a gamma distribution of site-rate variants across four categories. Trees were finally edited using iTOL v4. This model also was applied in Mega 6.0 to estimate nucleotide distances. Deduced amino acid sequence distances were estimated using the best-fitted model of Jones-Taylor-Thornton. In this analysis, a bootstrap procedure with 1,000 replicates was performed to estimate standard errors.

Potyvirus detection in the original locust tree
The presence of a potyvirus in the original locust tree (Robinia sp.) was tested. To this end, leaf samples were collected from this plant in the fall and spring of 2018 and 2019, respectively. Sampling was done from different branches of the plant and each sample was tested separately for potyvirus infections by enzyme-linked immunosorbent assay (ELISA) using potyvirus antibodies followed by RT-PCR using NIb primers, as above.
Results
Aphid transmission and serological assays
Virus-like symptoms consisting of necrotic dots, chlorotic patches and mottling, and discoloration, bleaching, mosaic patterns and necrotic ring spots appeared on leaves of two bean plants after inoculation with aphids collected from a Robinia sp. (locust) tree in the city of Tehran, i.e., mixed colonies of Aphis craccivora and A. fabae which were subsequently identified. The two virus-like isolates were respectively named Th-W2259 and Th-W2260 (Fig. 1). While systemic symptoms were induced for isolate Th-W2259, symptoms for the isolate Th-W2260 were limited to inoculated leaves. The identity of Th-W2259 as a potyvirus was confirmed by ELISA using potyvirus antibodies. Sample Th-W2260 had a negative reaction under ELISA for the same potyvirus antibodies.

(A) Apterous aphid species Aphis craccivora (left), and A. fabae (right), collected from a locust tree (Robinia sp.). (B) Virus-like symptoms induced on common bean (Phaseolus vulgaris) leaves inoculated by the aphids: isolate Th-W2259, left, and isolate Th-W2260, right.
RT-PCR, sequencing, and sequence analysis
The three universal primer pairs used in RT-PCR, i.e., CI, NIb, and NWCIEN primers, generated amplification products for the potyvirus isolate Th-W2259 with expected sizes of approximately 0.7-kb, 0.3-kb, and 0.7-kb, respectively (Ha et al., 2008; Valouzi et al., 2017; Zheng et al., 2010). Also, DNA fragments of about 1.0 kb were amplified using primers WVNIb1P and WVPseq designed to amplify the gap between NIb and CP regions. The sequences of the amplicons were determined. BLASTN analyses showed that the partial sequences of CI, NIb, and CP genes most closely matched sequences from WVMV isolates (genus Potyvirus), with pairwise sequence identity scores of 91%, 89%, and 99%, respectively. A contig consisting of the partial NIb, the complete CP and the partial 3′-untranslated region (3′UTR) was then assembled. The results showed that the complete CP gene of Th-W2259, composed of 846 nucleotides with deduced amino acid sequences of 282 aa, had the highest identities of 99% and 100%, respectively, to an Australian isolate of WVMV (accession no. AF484549). The genomic sequences were assigned GenBank accessions MH558669 (CI region) and MH558668 (NIb to 3′UTR). Phylogenetic analysis was performed using the concatenated CI, NIb, and CP sequences of the isolate. The ML analysis is shown (Supplementary Fig. 1). In all the trees, the sequence of the Iranian isolate was placed as a sister group of WVMV and clustered with potyviruses in the BCMV subgroup with high bootstrap values, similar to those reported previously (Liang et al., 2006).
Sequence analysis of WVMV isolates
The nucleotide sequence of the CP gene of the isolate Th-W2259 and the other sequences of the virus (Table 1) were assessed for evidence of recombination using RDP 4.39. As no clear recombination events (P < 1 × 10−6) were found among the CP gene sequences studied, all the 14 non-recombinant sequences and one isolate of SMV (the outgroup) were utilized for phylogenetic analysis using ML and NJ algorithms; the ML tree is shown in Fig. 2. The trees obtained from both methods placed the sequences into five major groups: East Asian-1 to 3, North American and World with high bootstrap values. In all the trees, the Iranian isolate and the other six isolates from Europe and Oceania were clustered in World group. Six isolates from China and one isolate from United States were placed in East Asian-1 to 3 and North American group, respectively. While all the isolates belonging to East Asian and North American groups were obtained from Wisteria sinensis, most of World group isolates were isolated from different Wisteria species.
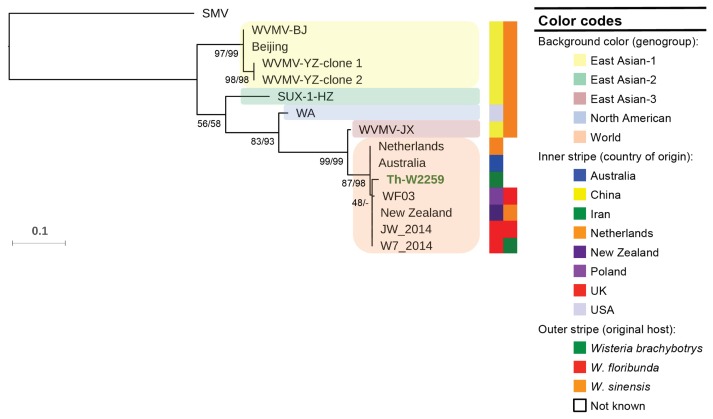
A maximum likelihood (ML) tree constructed from coat protein gene sequences of Wisteria vein mosaic virus isolates. Numbers at each node indicate the percentage of supporting bootstrap samples in ML and neighbor-joining methods, respectively. The homologous sequence of an isolate of soybean mosaic virus (accession no. FJ640971) was used as the outgroup. The sequence of the isolate Th-W2259 (in green) was obtained in this study.
Multiple sequence alignment showed high amino acid sequence similarities between the Iranian isolate, Th-W2259, and the other isolates. Although numerous conserved amino acid sequences were found among the isolates, especially among those belonging to the same phylogenetic group, a number of amino acid changes were observed among the isolates belonging to different groups. The results indicated that some of them were genogroup-specific amino acid substitutions, including A29, D14,88, E27,38, G2,125, K13,28, N20,35,258, Q3, R36, S18,19,34, T19, and V6. Moreover, the amino acid substitutions E125, I126, S120, and V74 were specific for World and East Asian-3 groups (Fig. 3).
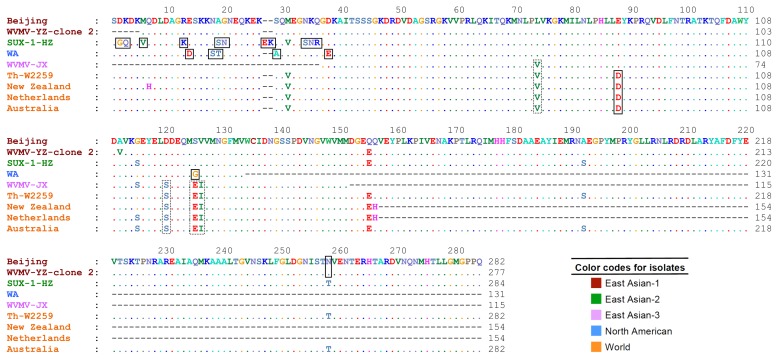
Amino acid sequence alignment of complete or partial coat protein sequences of representative isolates of Wisteria vein mosaic virus belonging to five phylogenetic groups: East Asian-1 to 3, North American and World. The sequence of the isolate Th-W2259 was obtained in this study. Amino acid substitutions are shown in black rectangles. Substitutions unique to the World and East Asian-3 groups are shown in dotted rectangles. The sequence of Beijing isolate was used as the reference sequence.
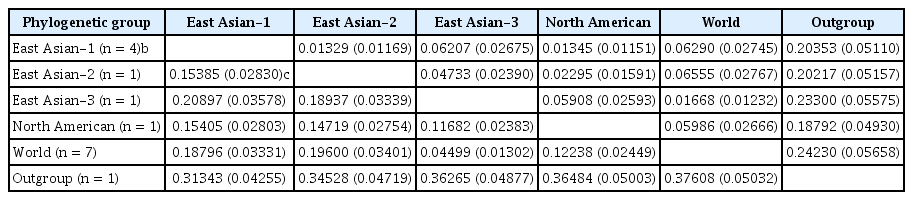
The net mean nucleotide distance between two groups, except for between the World and East Asian-3 groups, ranged from 0.12 to 0.21; the value obtained for the two groups was 0.04 (Table 2). So, the phylogenetic clustering of the isolates into at least four groups was reasonable. However, the East Asian-3 and World group may be either considered as two distinct groups, or one group namely World group. The mean nucleotide and amino acid sequence distances of the isolates in each group with respect to SMV sequence used as the outgroup were 0.31–0.38 and 0.19–0.24, respectively (Table 2). Nucleotide sequence comparisons between Chinese isolates, non-Chinese isolates, and between Chinese and other isolates were 0.00–0.22, 0.00–0.14, and 0.04–0.21, respectively. This value was 0.00–0.02 for the Iranian and other isolates from Oceania and Europe. Nucleotide and amino acid sequence distances of representative WVMV sequences from different genogroups are shown in Table 3. The overall nucleotide and amino acid sequence distances among the WVMV sequences were 0.12 and 0.02, respectively.

Potyvirus detection in the original locust tree
None of the leaf samples collected from the plant positively reacted in serological assays using the Potyvirus-specific antibodies. Negative reactions were also obtained in RT-PCR of the samples using NIb primers.
Discussion
In the present work, we tested whether aphids collected in the wild could be used as indicators of bean-infecting potyviruses. Individuals of A. craccivora and A. fabae collected from a locust tree transmitted a potyvirus to bean seedlings and produced symptoms resembled those reported for WVMV (Bos, 1996). A. craccivora was also previously reported as a vector for WVMV (Bos, 1996). Further studies, including sequencing and sequence analysis unequivocally confirmed the presence of the virus (Liang et al., 2006). Although we discovered a WVMV isolate from colonizing aphids collected from a locust tree, subsequent tests on the apparent host tree were negative for the virus. This could be due to uneven distribution of viruses in plants (Hull, 2014; Jones et al., 2017). Another interpretation of these results is that we accidentally collected a viruliferous winged aphid(s) from the locust tree. Winged aphids are mobile, and they may probe plants as they move and seek suitable food sources. As they probe, potyviruses adhere to mouthparts via motifs in the viral HC-Pro and CP (Whitfield et al., 2015). In this case, although the winged aphid was probably collected from a legume by chance, it is likely that it picked up WVMV from an earlier visit to another legume. Potyviruses remain viable and infectious in the mouthparts of aphids for a few hours (Fereres and Raccah, 2015), which results in spread of these viruses to new host plants. This finding highlights both a potential advantage and potential challenge to using aphids to sample the potyvirome of the area, especially when dealing with winged aphids.
The discovery of WVMV was surprising because this virus had not previously been reported from Iran or the rest of the western Asian region. However, we have not any information about the occurrence, distribution and the possible economical importance of the virus in different crops, e.g. legumes, in different geographical regions of the country. Phylogenetic analyses of WVMV isolates indicate five geographical subpopulations: three from China (East Asian-1 to 3), one from USA (North American) and one from Oceania, Europe and Iran (World) (Fig. 2); however, East Asian-3 and World groups may be also considered as one genogroup (Table 2). Similarly, the trees obtained from amino acid sequences categorized the sequences into three major groups: one from China (containing East Asian-1&2 isolates), one from USA (containing North American isolate) and one from Oceania, Europe, China, and Iran (containing World and East Asian-3 isolates) (figure not shown). The results seem to indicate that “geographical origin” may play an important role in the evolution of this virus; similar results have been obtained for other RNA viruses (Golnaraghi et al., 2018; Yasaka et al., 2017). While very low values (0.00–0.02) were obtained in nucleotide sequence comparisons between the isolates in the World group, high values (0.00–0.22) were calculated in pairwise comparisons between Chinese isolates, and between Chinese and non-Chinese isolates (Table 3). These findings, together with the results of the phylogenetic analyses, show a possible Eastern-Asian origin of the virus, because highest genetic diversity of a pathogen typically occurs in its center of origin (Jones, 2005). This region is considered as the main region where Wisteria originated (Li et al., 2014). Further studies seem necessary to confirm this hypothesis. It is worth noting that WVMV is limited to Wisteria species (Clover et al., 2003), and Robinia sp. is not listed as a host. Moreover, there are no wild Wisteria species in Iran; the ornamental species cultivated is W. sinensis (Mozaffarian, 2013). Therefore, the simplest explanation for the occurrence of this virus in Iran is that it was transported there by humans in Wisteria plants. The role of human in dissemination of other potyviruses has been suggested earlier (Yasaka et al., 2017). Virus-infected plants were possibly imported to Iran from Europe or Oceania; some of the countries in these regions have a lot of agricultural trades with Iran. This study also showed some genogroup-specific amino acid substitutions that can be used to design genogroup-specific primer pairs for fast and accurate detection of different populations of the virus, which can improve our future quarantine programs.
A limitation of the approach used in this study is the difficulty in identifying viruses that do not infect the experimental host plants. To solve this problem, a broad range of indicator plants or those susceptible to a wide range of plant viruses, e.g., Nicotiana benthamiana (Brunt et al., 1996), can be applied. The other issue happens when we are dealing with viruses for which genus or family-specific primers do not exist. This is illustrated in the challenges we are having to identify the second virus discovered here. HTS can be an effective tool for detecting unknown viruses because no previous knowledge of the virus sequence is required, and this approach has been used to identify known and novel viruses from wild and domesticated plants (Ong et al., 2017; Wylie and Jones, 2012; Wylie et al., 2012, 2013). However, the effectiveness of an HTS approach to screening the virome of the area by sampling aphids relies on the proportion of aphids carrying viruses. Furthermore, this method seems not to be highly reliable where viruses or viroids are in low titer (Jo et al., 2018). The average of non-persistent virus particles transmitted by an aphid was estimated to be as low as 0.5–3.2 (Moury et al., 2007). These issues and the transient nature of non-persistently transmitted plant viruses may be the main reasons for the failure to detect such viruses in aphids by using HTS approach in previous studies (Feng et al., 2017; Wamonje et al., 2017), so it is not a definitive measure of the virome of the region.
‘Virome’ was a term first used to define the total viral component of a species or ecosystem (Wegley et al., 2007), but has since been used to describe the sum of the viral genomes present on the planet (Paez-Espino et al., 2016). When the term virome is applied to an individual, it generally refers to the viruses replicating within a host (Virgin, 2014). To distinguish those viruses replicating within a host and those viruses, especially non-replicative ones, that are using the organism as a vector to transport them to another host, herein we propose the term ‘vectorome.’ The term describes viruses carried by a viral vector that can be transmitted to non-homologous hosts, e.g., from aphids to plants, but do not replicate in vector cells in many cases. For example, Aphis glycines virus 2 (ApGlV2) systemically infects the soybean aphid, Aphis glycines, where it is vertically transmitted to progeny (Liu et al., 2016). It does not infect plants, and thus is not part of the aphid vectorome. On the other hand, the non-persistent (non-replicative) potyvirus SMV is a plant-infecting virus vectored by this aphid and is a component of its vectorome. Persistent (replicative) viruses such as tospoviruses that replicate in both vectors and plant hosts, are also components of the vectorome. In other word, all viruses present in a vector, e.g., an aphid, at any given moment constitute its virome, but only viruses capable of being transmitted to a non-homologous host constitute the vectorome. Non-replicative viruses, such as SMV and many other RNA-based plant viruses, may remain infectious in their vectors for periods of minutes to hours (Fereres and Raccah, 2015; Hull, 2014), and thus the vectorome is of a transient nature. We propose that this term be applied to all viruses transmitted by all vectors to non-homologous hosts.
Previous studies have shown the occurrence of different aphid species capable of transmitting potyviruses in several parts of Iran that experience different climate conditions (Hashemi, 2015). Therefore, an extension of this survey to the other areas of Iran and the use of different approaches seem justified to understand more about aphid-borne potyviruses and their epidemiology. This work introduces a simple but effective approach to sample potyviruses, which can be possibly applied for other vector-transmitted plant viruses. Utilizing such approaches will help us understand more about vector-borne viruses in the area, optimize diagnostic systems for their fast and accurate detection, and monitor them effectively to control them before causing epidemies.
Supplemental Materials
Acknowledgments
This work was supported by grants 92001005 and 92001006 from the Iranian Group for the Promotion of Science (IGPS). The authors are grateful to Ali Rezvani (former researcher, Iranian Research Institute of Plant Protection, Tehran) for identification of aphids, to Valiollah Mozaffarian (Research Institute of Forests and Rangelands, Tehran) and Marjan Diyanat (Department of Weed Science, Science and Research Branch of Islamic Azad University, Tehran) for identification of weeds and wild plants, and to Ahmadreza Golnaraghi (School of Chemical Engineering, University of Tehran) for image editing.
Notes
Electronic Supplementary Material
Supplementary materials are available at The Plant Pathology Journal website (http://www.ppjonline.org/).